TOC
Introduction
I wanted to do something different on this blog and go through something that’s less of my side-project, and more of my project project, i.e. PET pharmacokinetic modelling. In Part 4, I’ll cover how to process and model arterial blood data using the kinfitr package.
When making use of reference tissue modelling in PET, all the data comes from the PET image, and everything is comparatively straightforward. When using an arterial input function, we have parallel series of measurements. While the PET is running, we are collecting blood in parallel, and often using both continuous and manual sampling. With that blood, we are measuring blood radioactivity, and plasma radioactivity, and the parent (unmetabolised) fraction, and the blood-to-plasma ratio. And we use all of these, together, to derive the arterial input function (AIF).
At my previous institution, we have something of a tradition of using data in as raw a format as possible, and we would simply interpolate our curves. However, this depends greatly on the number of samples we record as well as the quality of those samples. Here I’ll go through the process of working with this data, and modelling it as we see fit.
The parts of this series are as follows:
If you want to mess around with these vignettes yourself without needing to install the package, you can fire up a binder containing everything by clicking the button below.
![]()
Packages
First, let’s load up our packages
#remotes::install_github("mathesong/kinfitr")
library(kinfitr)
library(tidyverse)
library(knitr)
library(cowplot)
library(mgcv)
library(ggforce)
theme_set(theme_light())Blood Processing
The first, and quite serious issue, is that of storage. Data is often stored in different ways between, or even within research groups, and this does not bode well for data sharing and the development of broadly-applicable tools. This issue is, however, essentially resolved by the introduction of PET BIDS, which contains all the information which is required in addition to the image file, stored in a highly structured manner. kinfitr can automatically read data structured in this manner and pull in all the knowledge it needs using the bids_parse_study() command, which produces a tibble with rows for petinfo, blooddata and tactimes.
The blooddata is a structure for storing the raw blood data, as well as how this data should be modelled, and how the final resulting interpolated curves should be created, called input objects, which are used for modelling. Of course, it is easiest to create blooddata objects using data in BIDS structure. If you are not so lucky, but you do have the raw data, you can create a blooddata object using the create_blooddata_components() command.
Within the pbr28 data within kinfitr, we don’t have a full BIDS dataset at hand, but I’ve included the columns which would be returned from parsing the data. I’ll first just cut down the size of the data to make it a little bit quicker to process.
data(pbr28)
pbr28 <- pbr28[1:4,]
pbr28 <- pbr28 %>%
select(-procblood, -input, -Genotype, -injRad, -tactimes, -petinfo)
head(pbr28)## # A tibble: 4 x 5
## # Groups: PET [4]
## PET Subjname PETNo tacs blooddata
## <chr> <chr> <dbl> <list> <list>
## 1 cgyu_1 cgyu 1 <tibble [38 x 10]> <blooddat>
## 2 cgyu_2 cgyu 2 <tibble [38 x 10]> <blooddat>
## 3 flfp_1 flfp 1 <tibble [38 x 10]> <blooddat>
## 4 flfp_2 flfp 2 <tibble [38 x 10]> <blooddat>Dispersion Correction
The first step is dispersion correction of blood samples collected through the automatic blood sampling system: as blood travels through the plastic tubing, it sticks to the sides, and there is some dispersion of our measured signal. This is corrected for using the dispersion constant (included in the BIDS sidecar). This is a value representing the degree to which our measurements are dispersed over time, based on experiments performed when calibrating the automatic blood sampling system. All functions which can be used on the blooddata objects are prefixed with bd_, in this case bd_blood_dispcor.
pbr28 <- pbr28 %>%
mutate(blooddata = map(blooddata,
~bd_blood_dispcor(.x)))In this case, we first perform dispersion correction. This makes the data slightly noisier, so we can also smooth the data slightly if we choose. I’ve not done so above, and would only do so if the data looked really nasty, and fits started failing.
The blooddata object
Inside the ´blooddata´ object is a complex list.
blooddata <- pbr28$blooddata[[3]]
str(blooddata)## List of 3
## $ Data :List of 3
## ..$ Blood :List of 2
## .. ..$ Continuous:List of 6
## .. .. ..$ Values : tibble [600 x 2] (S3: tbl_df/tbl/data.frame)
## .. .. .. ..$ time : num [1:600] 1 2 3 4 5 6 7 8 9 10 ...
## .. .. .. ..$ activity: num [1:600] 1.435 0.45 -0.029 -0.502 -0.118 ...
## .. .. ..$ DispersionConstant : num 2.6
## .. .. ..$ DispersionCorrected: logi TRUE
## .. .. ..$ Avail : logi TRUE
## .. .. ..$ time :List of 2
## .. .. .. ..$ Description: chr "Time in relation to time zero defined by the _pet.json"
## .. .. .. ..$ Units : chr "s"
## .. .. ..$ activity :List of 2
## .. .. .. ..$ Description: chr "Radioactivity in uncorrected whole blood samples from Allogg autosampler."
## .. .. .. ..$ Units : chr "kBq/ml"
## .. ..$ Discrete :List of 4
## .. .. ..$ Values : tibble [14 x 2] (S3: tbl_df/tbl/data.frame)
## .. .. .. ..$ time : num [1:14] 40 160 280 400 520 610 1180 1780 2380 2980 ...
## .. .. .. ..$ activity: num [1:14] 56.42 16.04 11.27 10.09 9.66 ...
## .. .. ..$ Avail : logi TRUE
## .. .. ..$ time :List of 2
## .. .. .. ..$ Description: chr "Time in relation to time zero defined by the _pet.json"
## .. .. .. ..$ Units : chr "s"
## .. .. ..$ activity:List of 2
## .. .. .. ..$ Description: chr "Radioactivity in whole blood samples."
## .. .. .. ..$ Units : chr "kBq/ml"
## ..$ Plasma :List of 4
## .. ..$ Values : tibble [14 x 2] (S3: tbl_df/tbl/data.frame)
## .. .. ..$ time : num [1:14] 40 160 280 400 520 610 1180 1780 2380 2980 ...
## .. .. ..$ activity: num [1:14] 46.21 11.02 8 7.43 9.18 ...
## .. ..$ Avail : logi TRUE
## .. ..$ time :List of 2
## .. .. ..$ Description: chr "No description"
## .. .. ..$ Units : chr "s"
## .. ..$ activity:List of 2
## .. .. ..$ Description: chr "Radioactivity in plasma samples."
## .. .. ..$ Units : chr "Time in relation to time zero defined by the _pet.json"
## ..$ Metabolite:List of 6
## .. ..$ Values : tibble [9 x 2] (S3: tbl_df/tbl/data.frame)
## .. .. ..$ time : num [1:9] 0 60 180 300 630 1200 2400 3600 5400
## .. .. ..$ parentFraction: num [1:9] 1 0.996 0.921 0.706 0.263 ...
## .. ..$ Avail : logi TRUE
## .. ..$ time :List of 2
## .. .. ..$ Description: chr "Time in relation to time zero defined by the _pet.json"
## .. .. ..$ Units : chr "s"
## .. ..$ parentFraction :List of 2
## .. .. ..$ Description: chr "Parent fraction of the radiotracer."
## .. .. ..$ Units : chr "unitless"
## .. ..$ Method : chr "HPLC"
## .. ..$ RecoveryCorrectionApplied: logi TRUE
## $ Models :List of 4
## ..$ Blood :List of 2
## .. ..$ Method: chr "interp"
## .. ..$ Data : NULL
## ..$ BPR :List of 2
## .. ..$ Method: chr "interp"
## .. ..$ Data : NULL
## ..$ parentFraction:List of 2
## .. ..$ Method: chr "interp"
## .. ..$ Data : NULL
## ..$ AIF :List of 2
## .. ..$ Method: chr "interp"
## .. ..$ Data : NULL
## $ TimeShift: num 0
## - attr(*, "class")= chr "blooddata"Essentially, it can be divided into a few parts.
names(blooddata)## [1] "Data" "Models" "TimeShift"At the first level of the hierarchy, we have timeshift numeric value, which is there in case we know that our time values have been shifted by some amount. Within Data, we have most of the PET BIDS blood fields from the JSON sidecar. And within Models, we have the methods by which the blood data should be interpolated to created the final interpolated curves for modelling. In this case, because we have not yet done anything, everything is simply linearly interpolated.
blooddata$Models## $Blood
## $Blood$Method
## [1] "interp"
##
## $Blood$Data
## NULL
##
##
## $BPR
## $BPR$Method
## [1] "interp"
##
## $BPR$Data
## NULL
##
##
## $parentFraction
## $parentFraction$Method
## [1] "interp"
##
## $parentFraction$Data
## NULL
##
##
## $AIF
## $AIF$Method
## [1] "interp"
##
## $AIF$Data
## NULLWithin these, the Method field describes what should be done, and the Data fields describes what should be used for doing that. Because of the fact that there are loads of different ways to model blood data, and because many of these ways are defined in an ad hoc manner, I’ve designed the system to be as flexible as possible. We can add:
- Model parameters for models which are defined within kinfitr (
bd_addfitpars()) - Fit objects for models we can define using R, but which aren’t necessarily in kinfitr (
bd_addfit()). I tend to find this approach most useful, even for methods within kinfitr. - Fitted values for methods which exist outside of R, or do not function at an individual level (
bd_addfitted()).
Once we’ve done all the modelling of the blood with the blooddata object, we can interpolate it into an input object that we used in Part 2 of the tutorial.
Modelling the Blood
Let’s have a look at a few blood curves
plot(pbr28$blooddata[[1]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).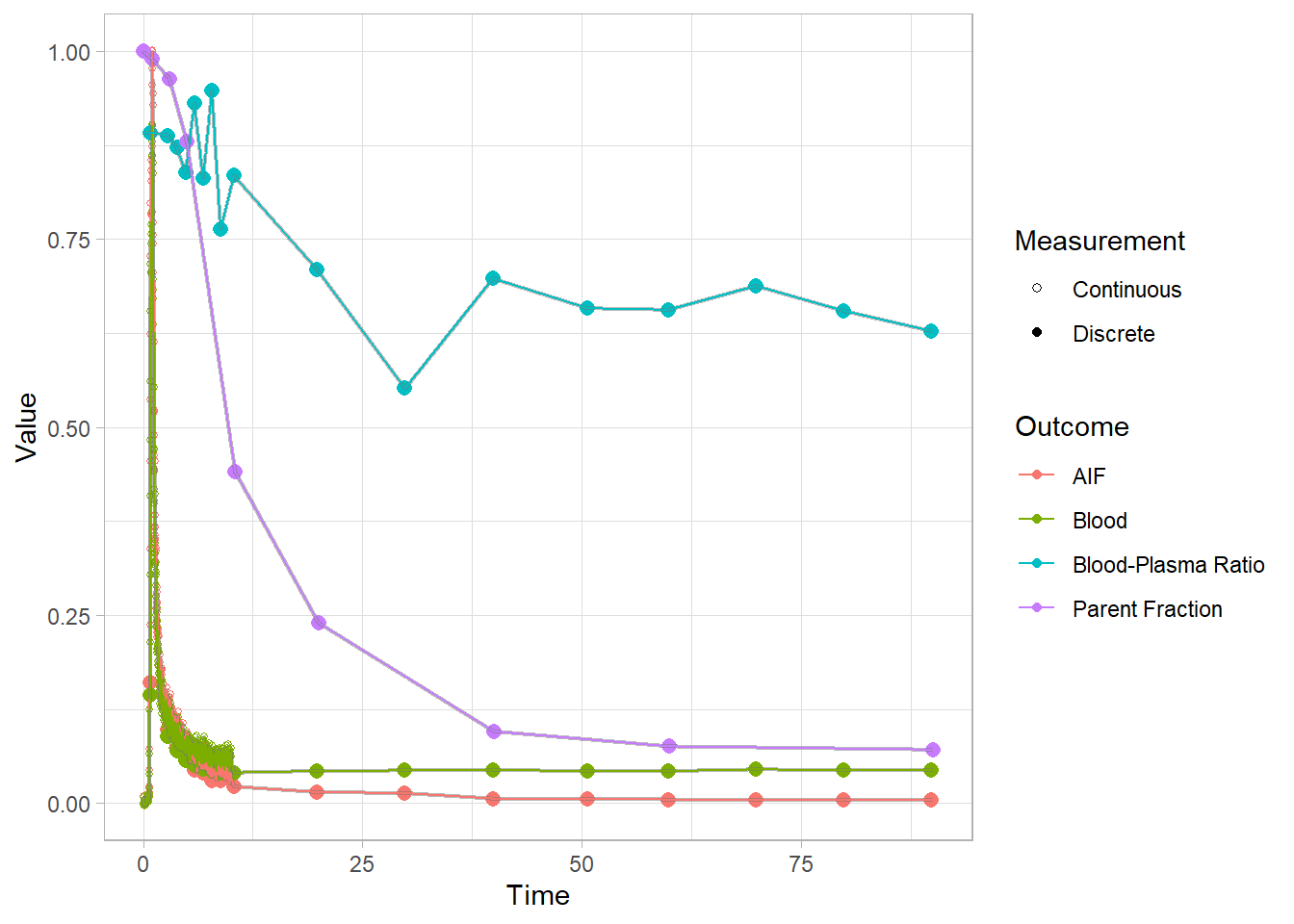
plot(pbr28$blooddata[[2]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).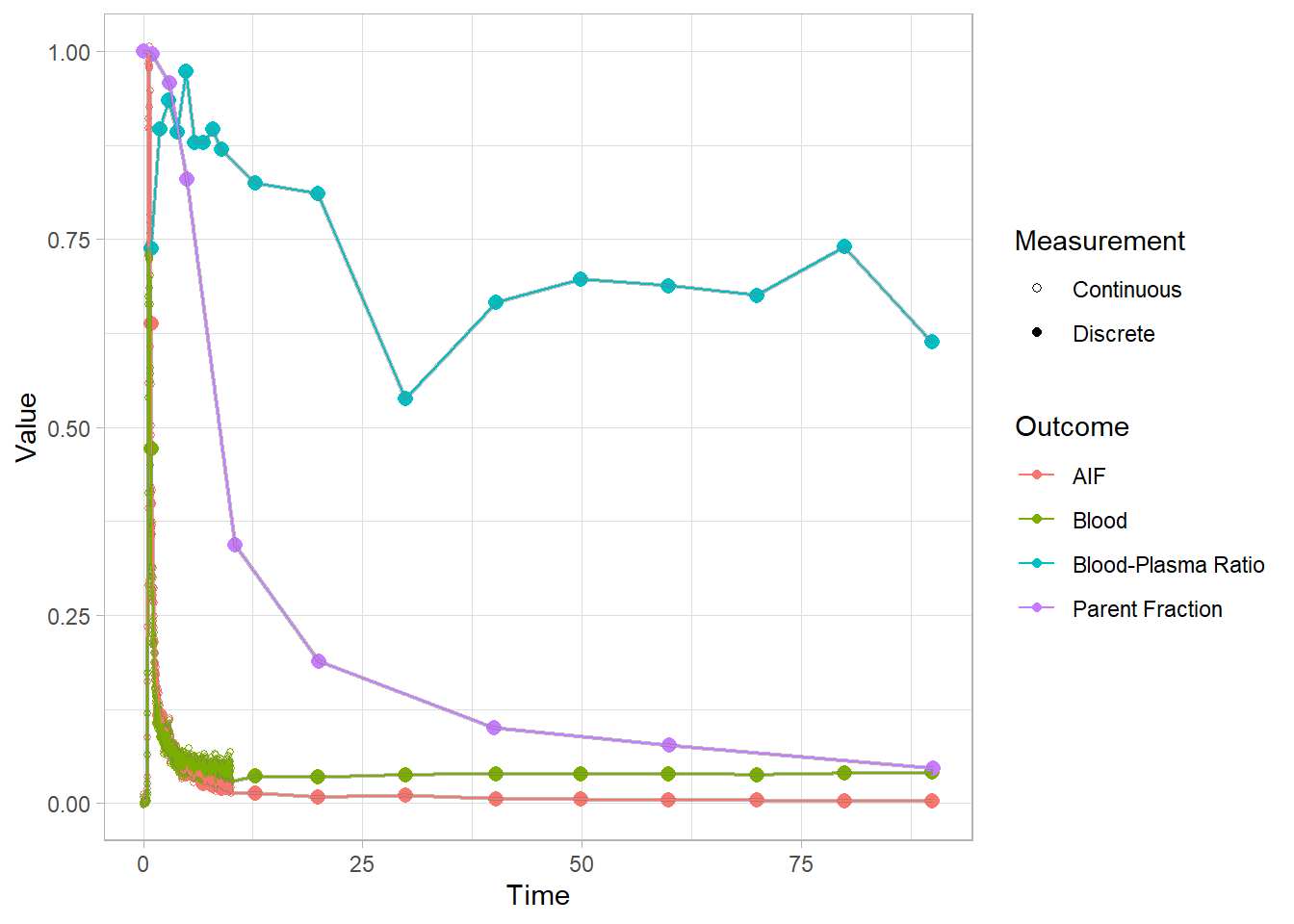
plot(pbr28$blooddata[[3]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).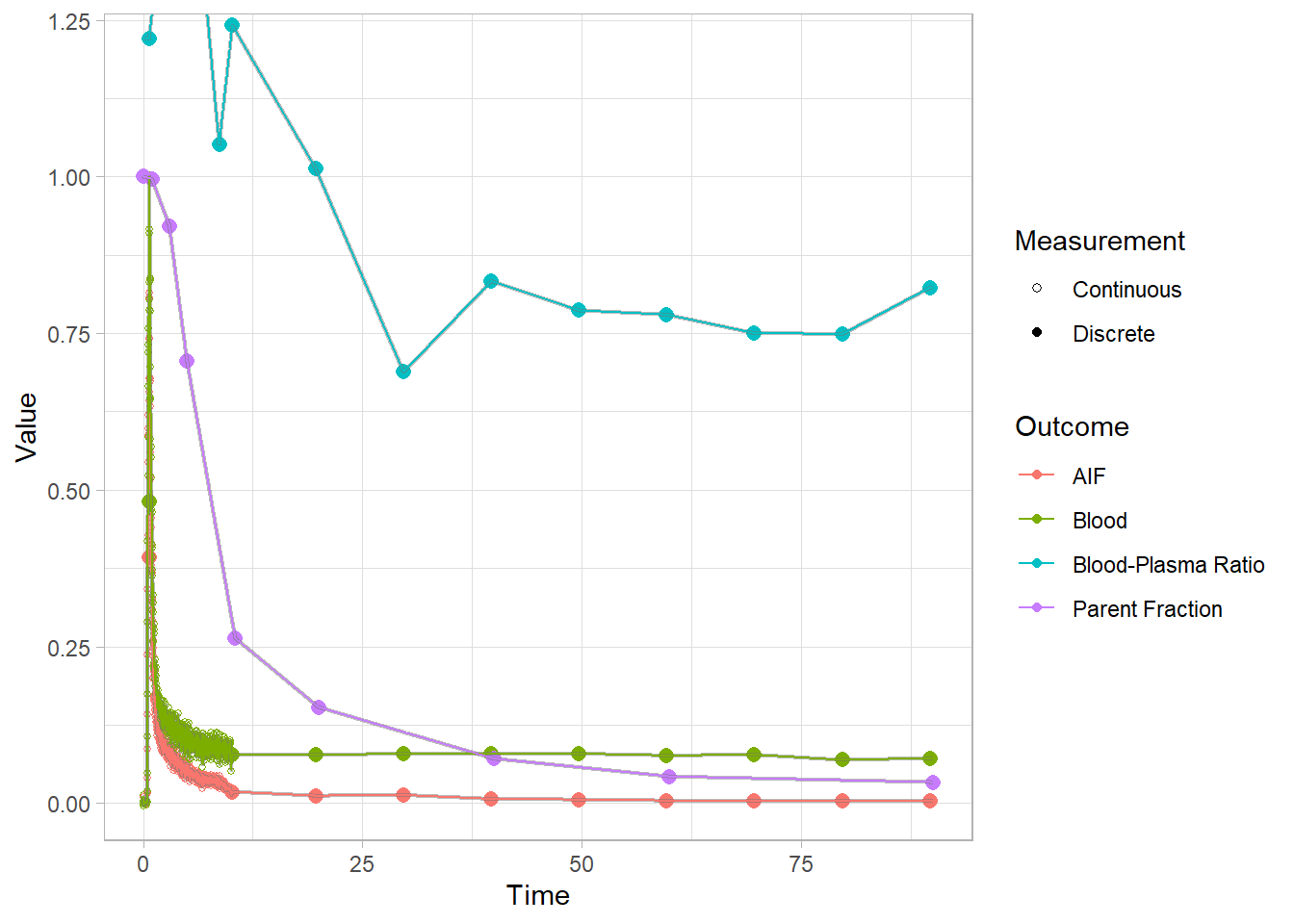
For these images, I opted to re-scale the blood and AIF curves (scaled to a shared maximum value, so they’re on the same scale) so that we can see all the curves together. This is because the final AIF is estimated based on all of the other curves, and if something is wrong, it’s helpful to see how and where it could be affecting the rest, not least because we often have to rely on other modelling steps for fitting later modelling steps.
Let’s model some of these curves to make them a bit smoother.
Modelling the Parent Fraction
Within kinfitr, there are a few different models for the parent fraction, which can be used at will, and compared easily using various model comparison metrics.
Here I’ll try out a few, and we can see how they do. The Guo modification of the Hill function was specifically designed for tracers within the same family as PBR28, so we expect it to perform especially well.
To get data out of the bloddata object, we use the bd_getdata() function, and then we specify what we want.
Note that all models for the parent fraction, start with metab_ so that they can easily be shuffled through.
pfdat <- bd_extract(blooddata, output = "parentFraction")
powermodel <- metab_power(pfdat$time, pfdat$parentFraction)
expmodel <- metab_exponential(pfdat$time, pfdat$parentFraction)
invgammamodel <- metab_invgamma(pfdat$time, pfdat$parentFraction)
hillmodel <- metab_hill(pfdat$time, pfdat$parentFraction)
hillguomodel <- metab_hillguo(pfdat$time, pfdat$parentFraction)
BIC( powermodel, expmodel, invgammamodel, hillmodel, hillguomodel ) %>%
rownames_to_column("Model") %>%
as_tibble() %>%
arrange(BIC)## # A tibble: 5 x 3
## Model df BIC
## <chr> <dbl> <dbl>
## 1 hillguomodel 4 -39.9
## 2 powermodel 4 -39.8
## 3 hillmodel 4 -35.8
## 4 invgammamodel 3 -32.8
## 5 expmodel 4 -11.6According to the BICs, our HillGuo model appears to be performing best.
Let’s look at some plots. We’ll add the fits to the blooddata object, and plot them there. We’ll be looking at the purple line.
bd_addfit(blooddata, powermodel, modeltype = "parentFraction") %>%
plot() + labs(title="Power Model")## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).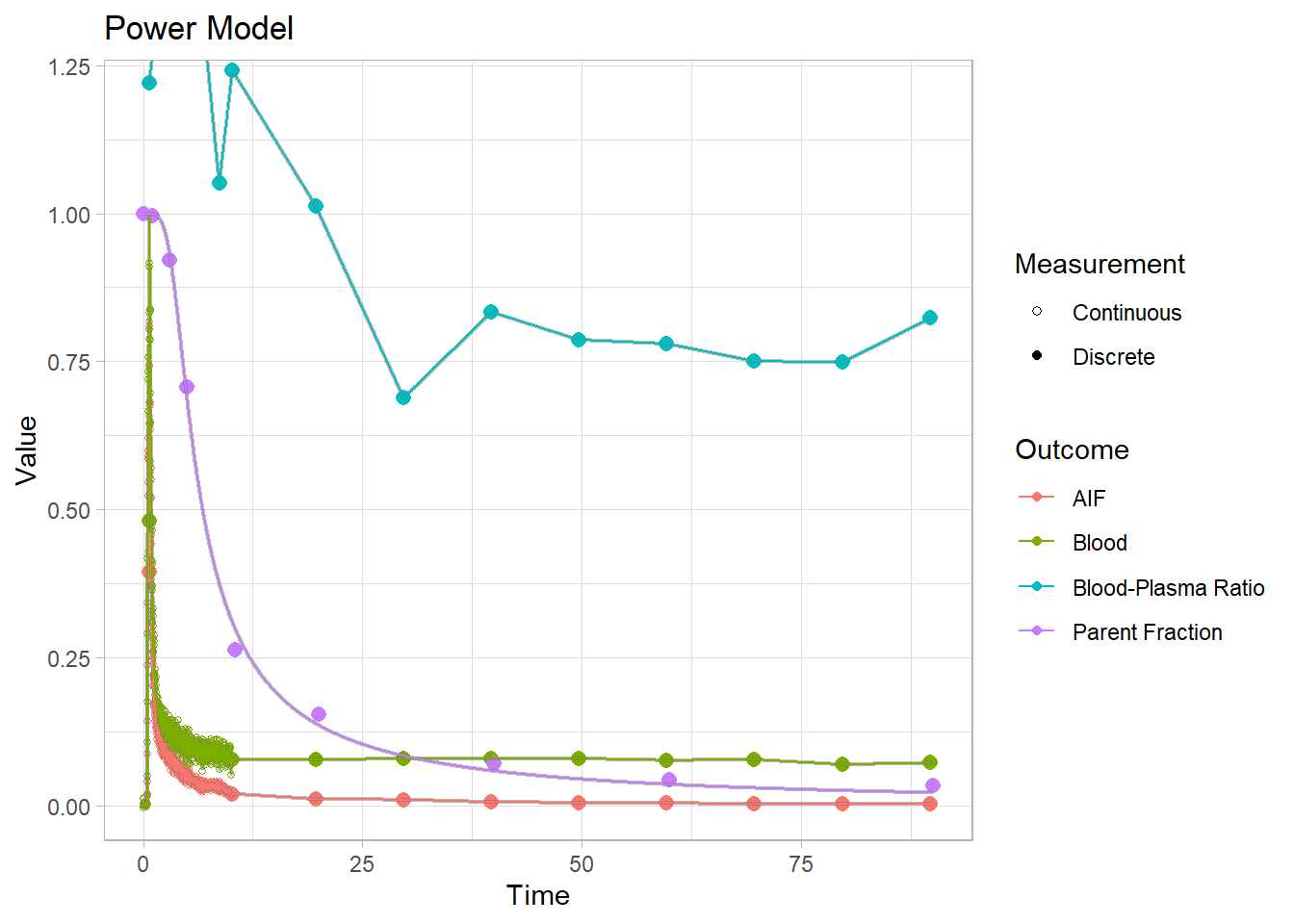
bd_addfit(blooddata, expmodel, modeltype = "parentFraction") %>%
plot() + labs(title="Exponential Model")## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).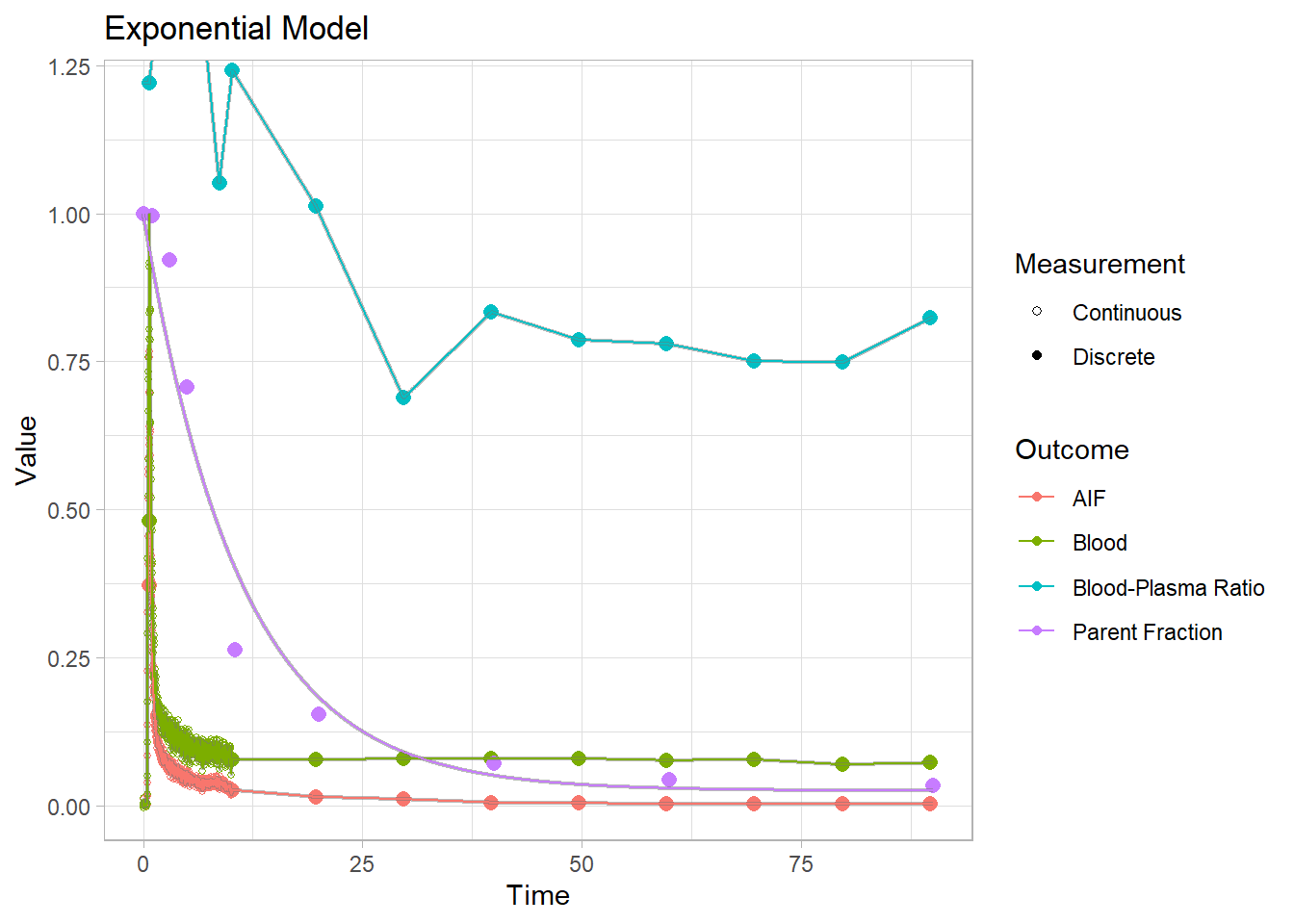
bd_addfit(blooddata, invgammamodel, modeltype = "parentFraction") %>%
plot() + labs(title="Inverse Gamma Model")## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).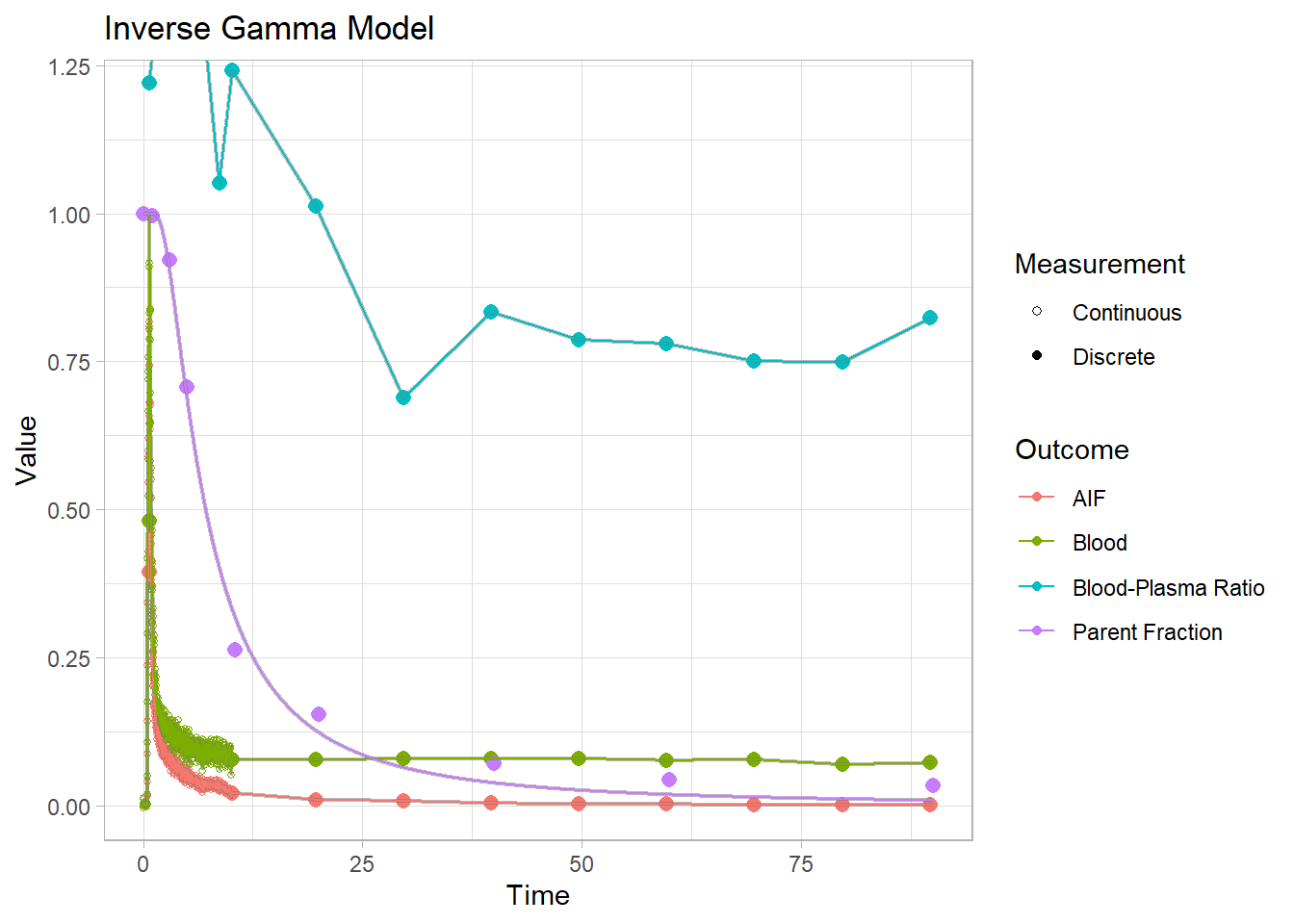
bd_addfit(blooddata, hillmodel, modeltype = "parentFraction") %>%
plot() + labs(title="Hill Model")## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).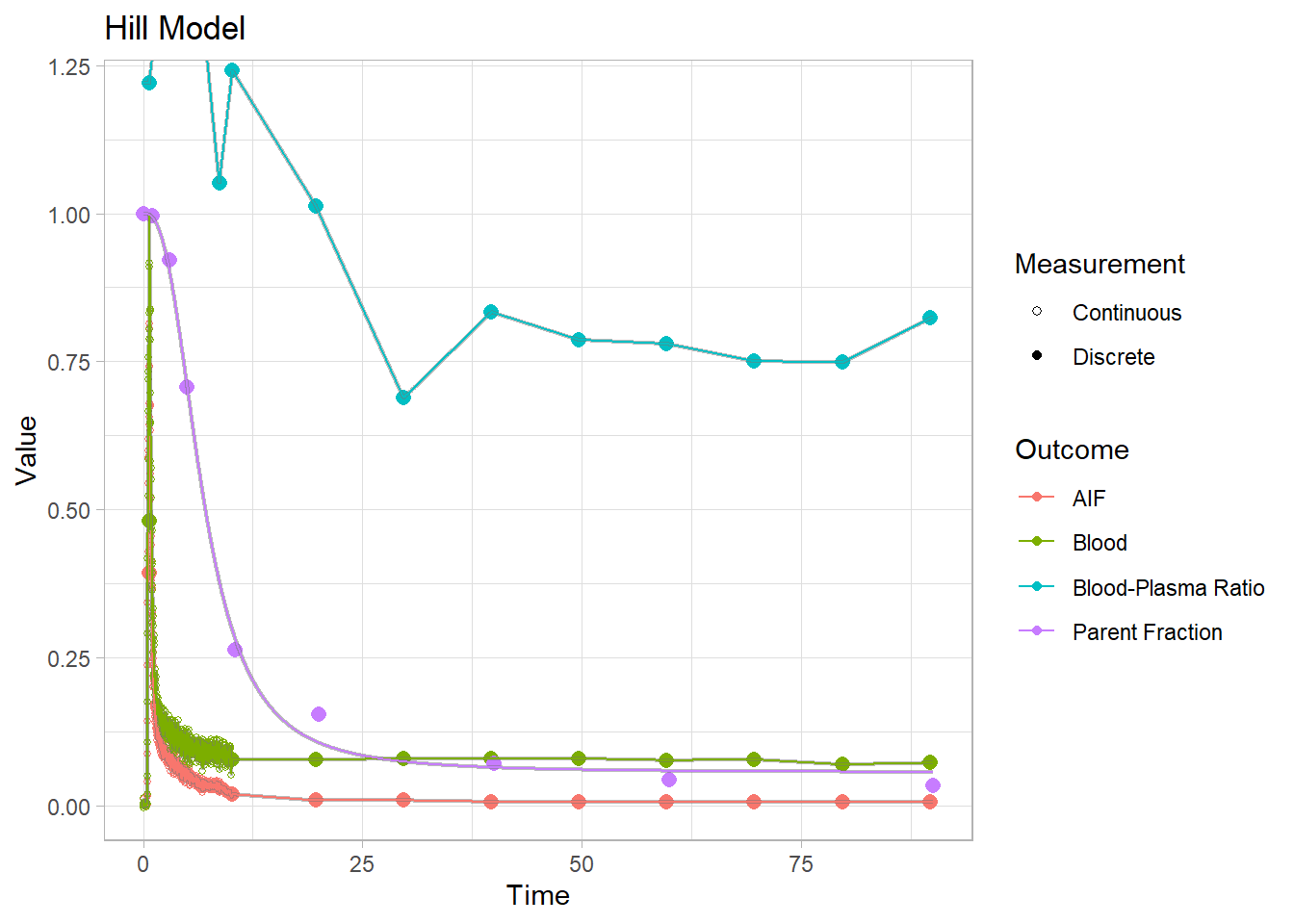
bd_addfit(blooddata, hillguomodel, modeltype = "parentFraction") %>%
plot() + labs(title="Guo Modification of Hill Model")## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).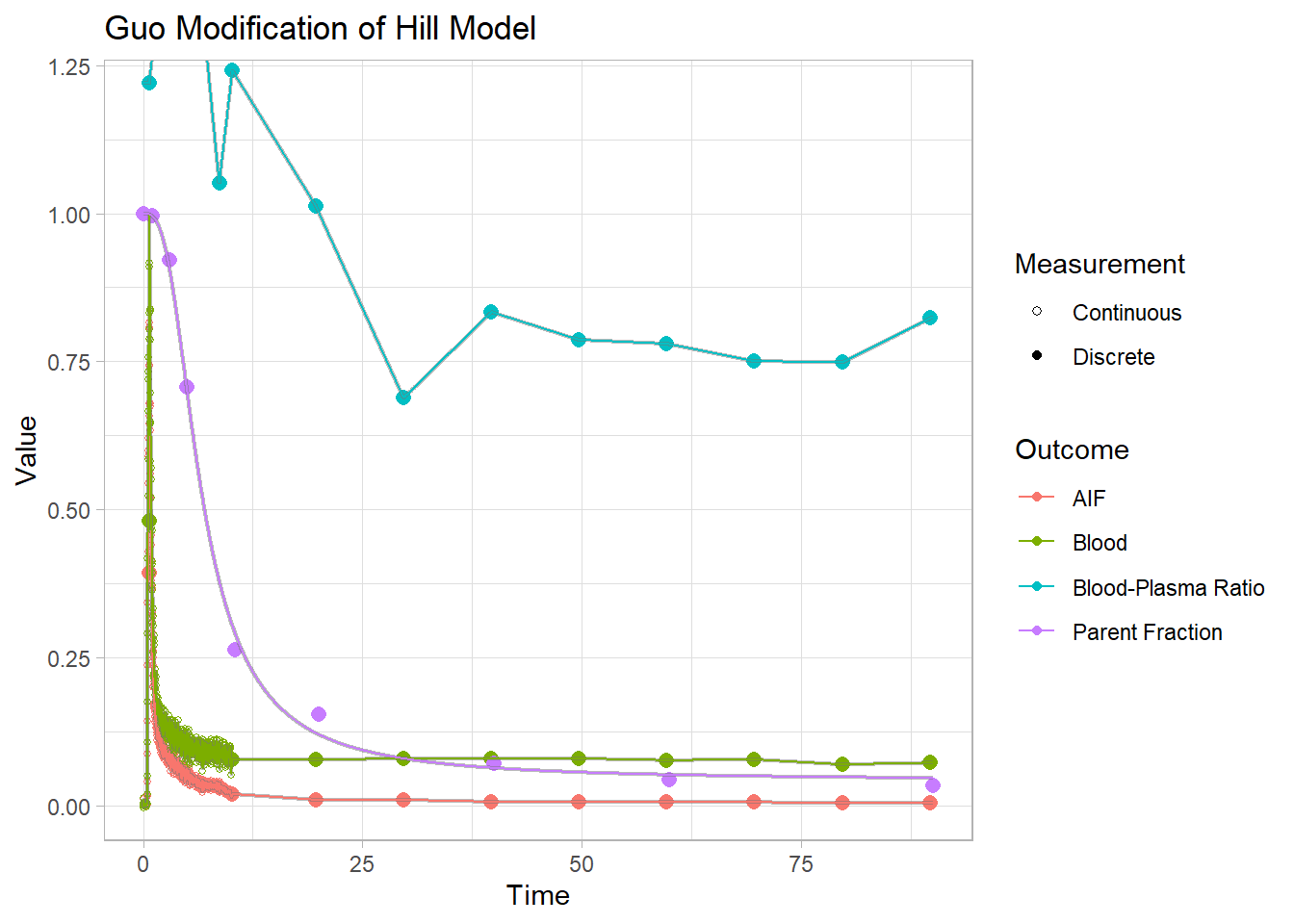
Each of these models have their own specific shapes, which we must select between to model the shape of the metabolism curve for each tracer. And then there are heaps of modification of all the different models which are created to adapt to curves which slightly differ from the primary models. In this case, we can see that the power and inverse gamma models are not able to level out towards the end sufficiently. The exponential model can’t handle the slow start, but does level out. The Hill model handles the end nicely, but can’t quite hit the values in the middle. The Guo modification does a pretty good job of representing the curve though.
Obviously, we can’t make these generalisations from just one curve, but if you would look at the rest, you would see that these biases at various parts of the curve are quite consistent between the different curves. I usually encourage performing model comparison with BIC and AIC and seeing which models are winning most of the time, and then looking at a few plots to get a feeling for why they’re winning out.
I’ll proceed with the hillguo model for demonstration purposes (which ), and add it to all our blooddata objects.
set.seed(123)
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(pfdat = map(blooddata,
~bd_extract(.x, output = "parentFraction")),
hillguomodel = map(pfdat,
~metab_hillguo(.x$time, .x$parentFraction, multstart_iter = 100)),
blooddata = map2(blooddata, hillguomodel,
~bd_addfit(.x, .y, modeltype = "parentFraction"))) %>%
select(-pfdat, -hillguomodel)Let’s check out how it went for a few. In proper analyses, I would advise looking at all fits, because these can go wrong. And if they do go wrong, they can often be fixed by trying to fit them again: these functions use a multstart approach, because the starting parameters can differ so greatly from tracer to tracer that they’re a pain to define. You can use more iterations, or a new seed. (PS: I also recommend always setting the seed first).
plot(pbr28$blooddata[[1]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).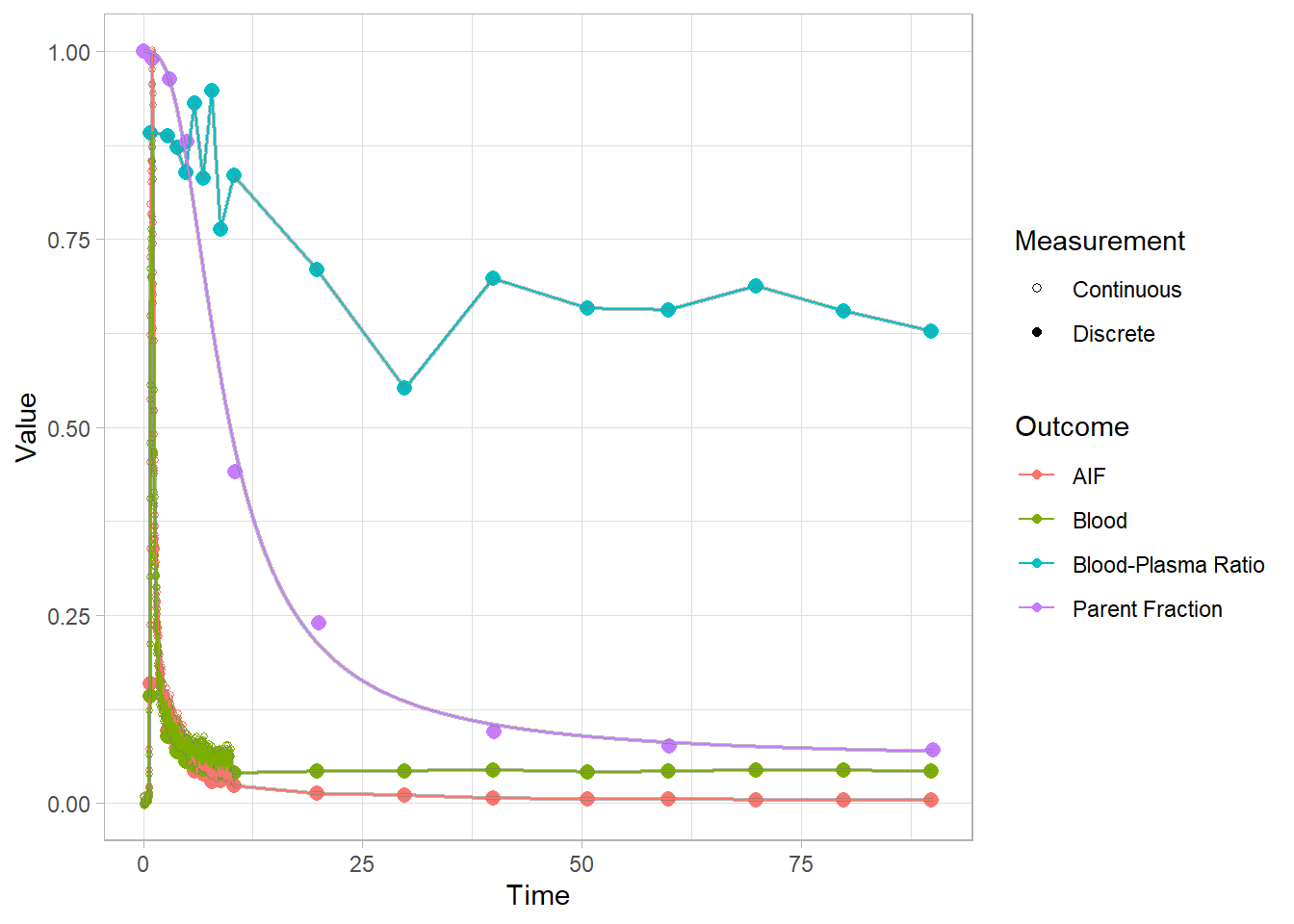
plot(pbr28$blooddata[[2]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).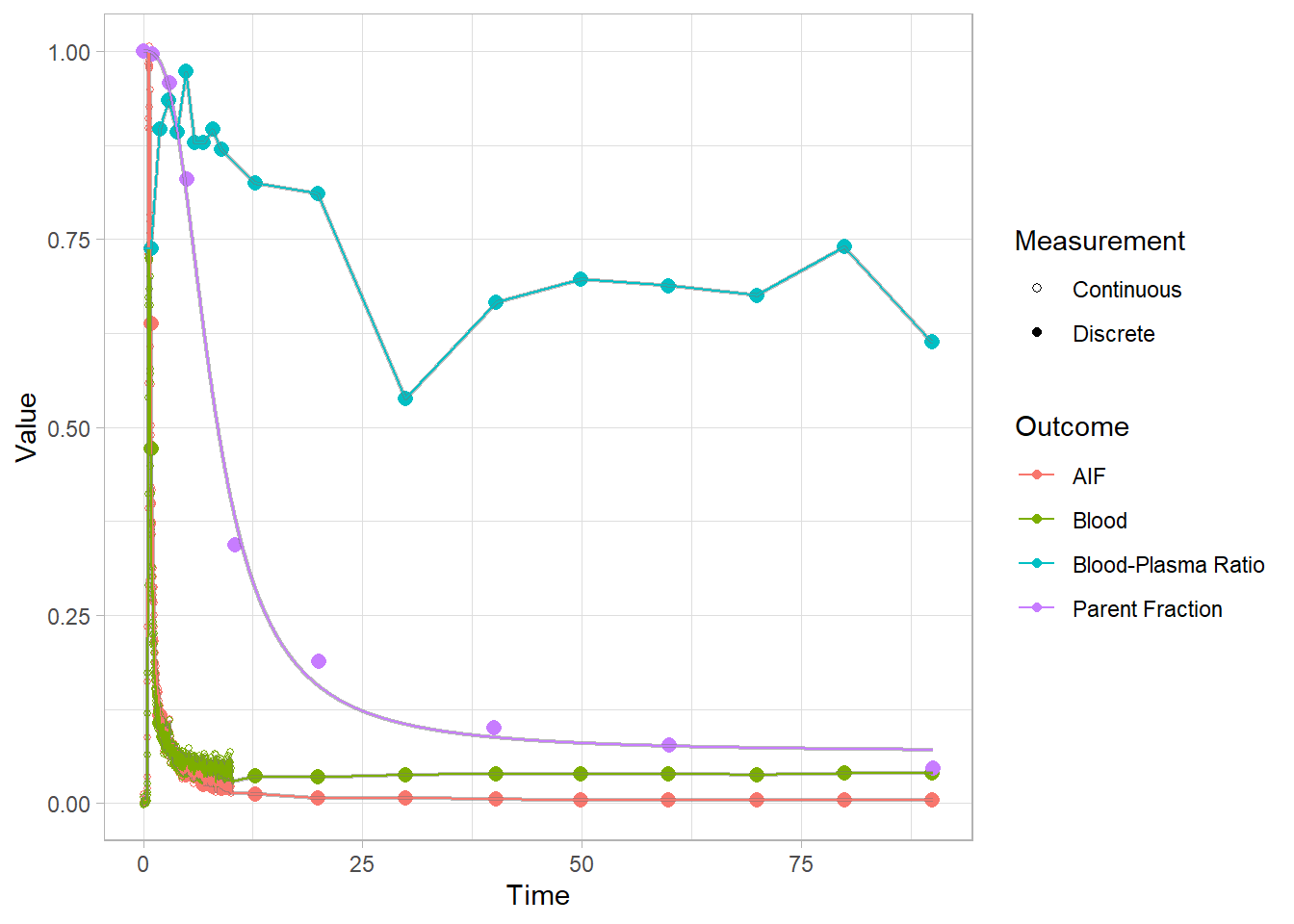
plot(pbr28$blooddata[[3]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).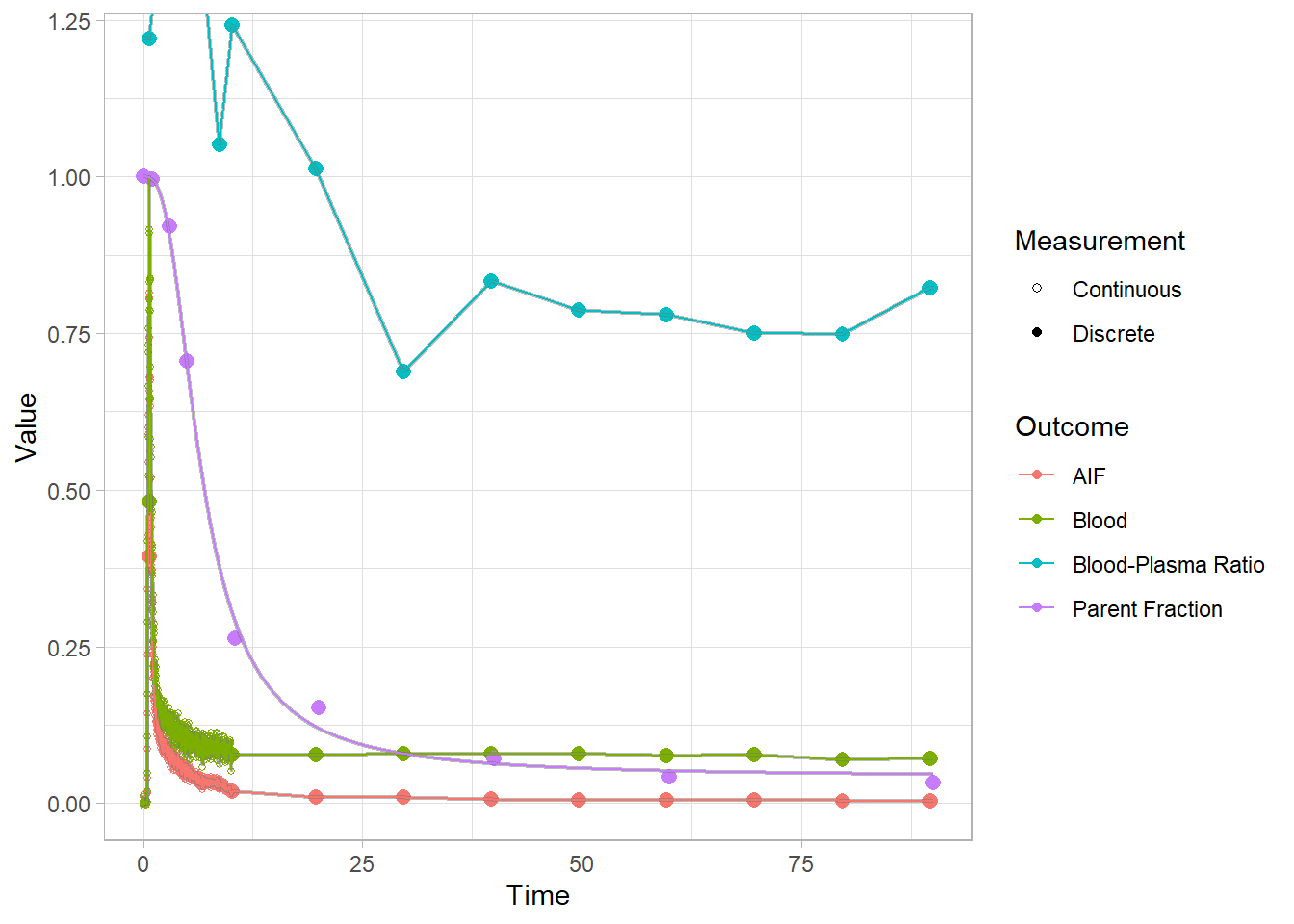
Now we see a much smoother fit to the parent fraction. This will surely do a better job of representing this curve than a simple interpolation. However, we do also see that we consistently underestimate around 20 minutes. We’ll just have to live with this…
Modelling the blood-plasma ratio
The blood-plasma ratio doesn’t seem to follow any particular function here, and there are no in-built methods for modelling this in kinfitr. Sometimes, it is constant. Sometimes it can be modelled with a straight line. Sometimes with a particular function. In this case, we see that it just appears really wonky. This could either be because it shows oddd kinetics, or because it’s hard to measure. This will probably benefit from a smooth line fit through it. In R, we can do this using a Generalised Additive Model (GAM). That’s not built into kinfitr, but let’s try it in R, and just throw that into the blooddata object. We’ll do it for everyone at once.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(bprdat = map(blooddata,
~bd_extract(.x, output = "BPR")),
bprmodel = map(bprdat,
~gam(bpr ~ s(time), data=.x)),
blooddata = map2(blooddata, bprmodel,
~bd_addfit(.x, .y, modeltype = "BPR"))) %>%
select(-bprdat, -bprmodel)And let’s check how the plots look. Here we’re looking at the blue curves at the top.
plot(pbr28$blooddata[[1]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).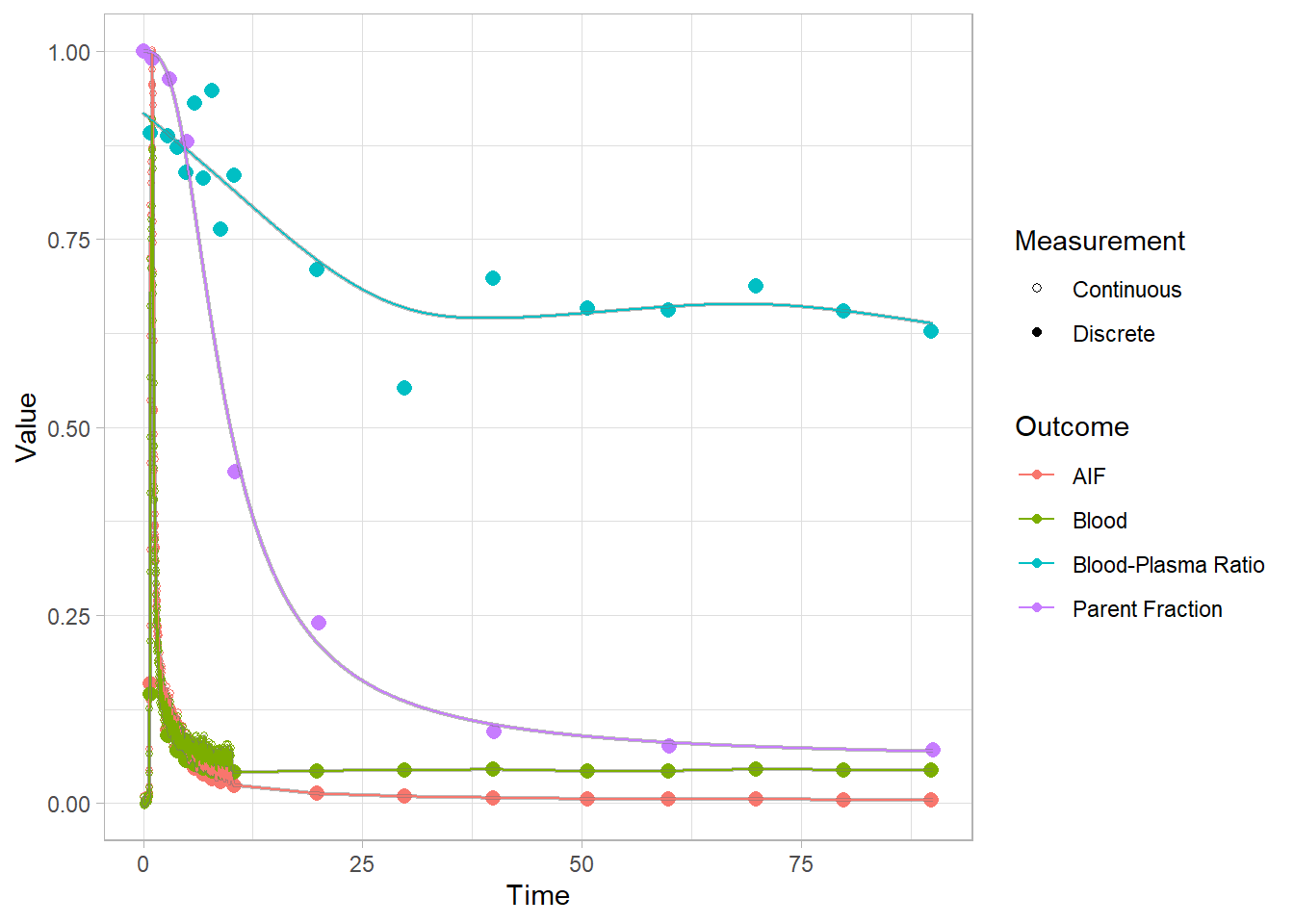
plot(pbr28$blooddata[[2]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).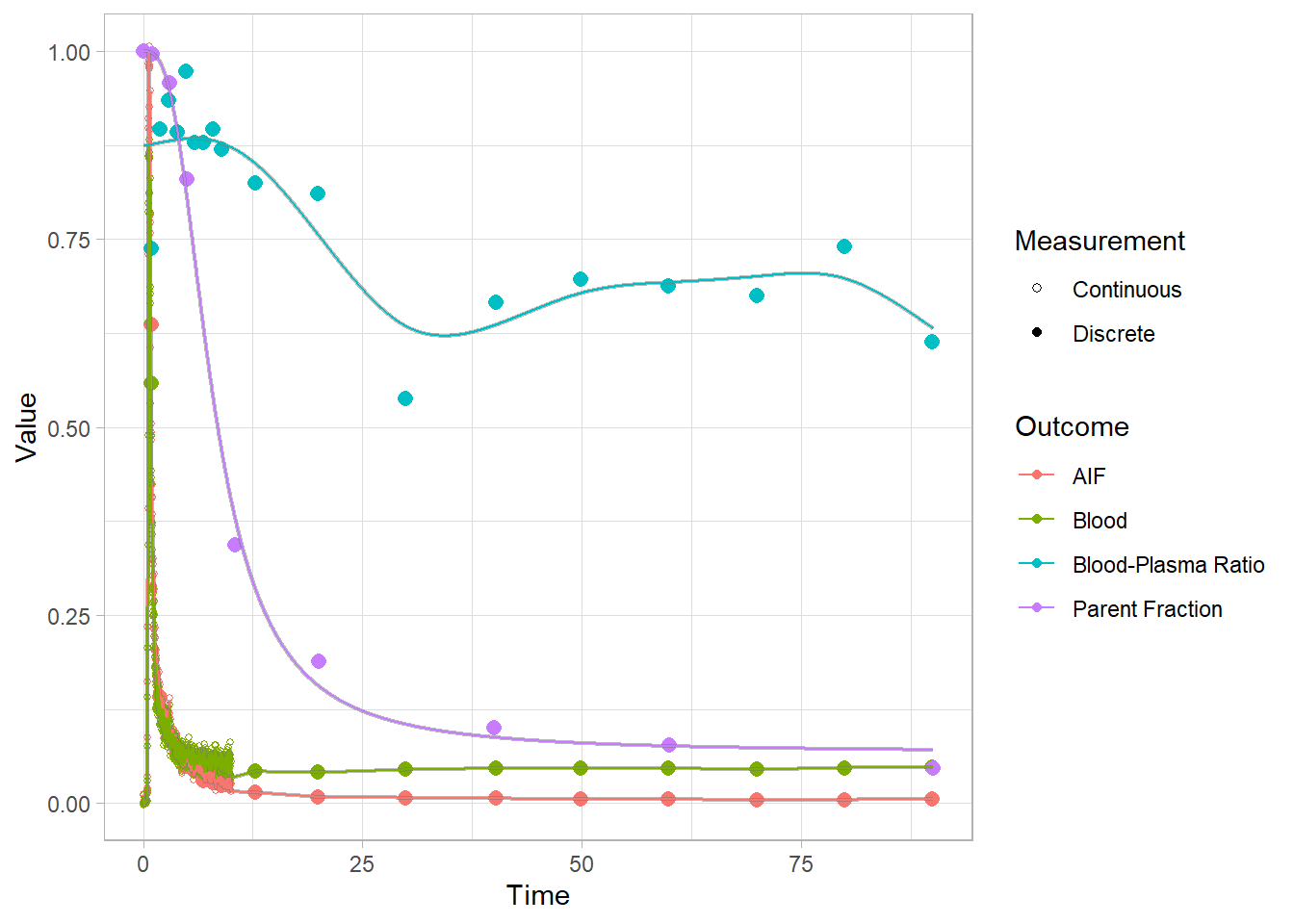
plot(pbr28$blooddata[[3]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).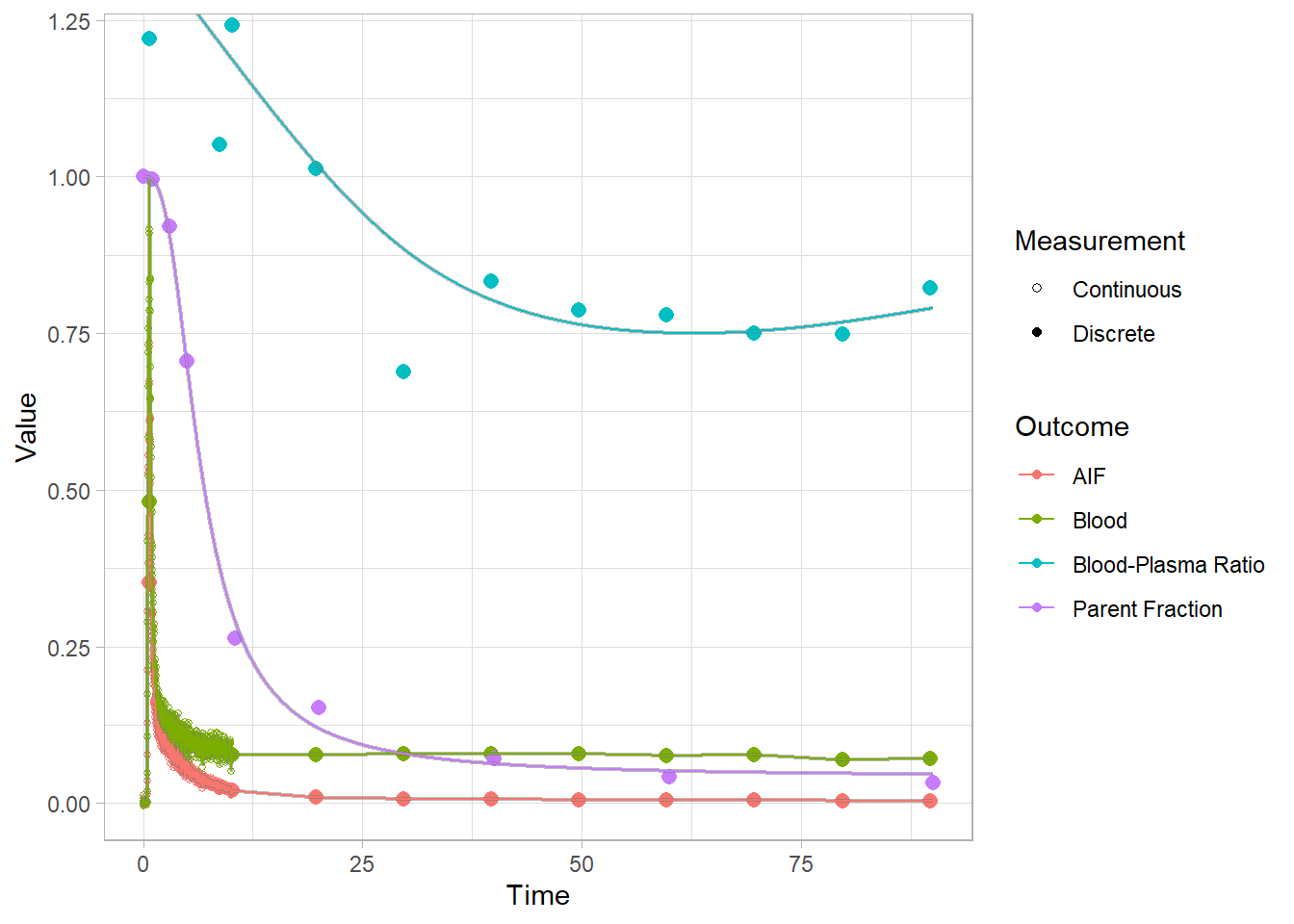
Clearly the fits are not fantastic, but I certainly trust them more than before. An interpolation would be going all over the show here, so I think we’ll settle for these.
Modelling the blood and arterial input function
Let’s also go ahead and model the blood and AIF too while we’re at this. It’s important to note that I do this last of all. This is because the AIF ‘data’ that we have, is the result of the model fitting we’ve performed above. And for the blood, we’ve modelled the blood-to-plasma ratio (BPR) using the raw data. We will use this curve then to apply to the blood curve that we have, which we would ideally like to smooth a little bit.
Blood
Modelling the blood specifically is not so common: it can be tricky. But it is helpful for us to know how much of the radioactivity in the brain is attributable to the radioactivity which is just in the blood which is passing through (and makes up about 5% of the tissue). It is especially tricky though, because this curve can go up towards the end sometimes, so many functions can’t adequately fit it. Furthermore, when we have both continuous and manual samples, we have an extremely high density of samples at the start and much fewer towards the end.
For modelling of blood or AIF data, there is a spline function in kinfitr, which fits three splines: one to the rise, one to the automatic fall, and one to the manual fall. And then we gradually hand off the automatic to the manual fall. This tends to work quite well in my experience. In this particular study, there were some issues with the blood data collection, and it is believed that the automatic sampling system was placed slightly too near to the participants for some measurements, and so there is an offset between the manual and automatic samples, and the hand-off method handles this quite well. Let’s give it a shot.
Note: if you are going to model your AIF, I would advise either not modelling the blood, or else modelling it after the AIF. This will become clear later. I’m doing it here to make something else clearer later, for pedagogical reasons.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(bddat = map(blooddata,
~bd_extract(.x, output = "Blood")),
bdmodel = map(bddat,
~blmod_splines(.x$time, .x$activity, Method = .x$Method)),
blooddata = map2(blooddata, bdmodel,
~bd_addfit(.x, .y, modeltype = "Blood"))) %>%
select(-bddat, -bdmodel)Let’s see
plot(pbr28$blooddata[[1]])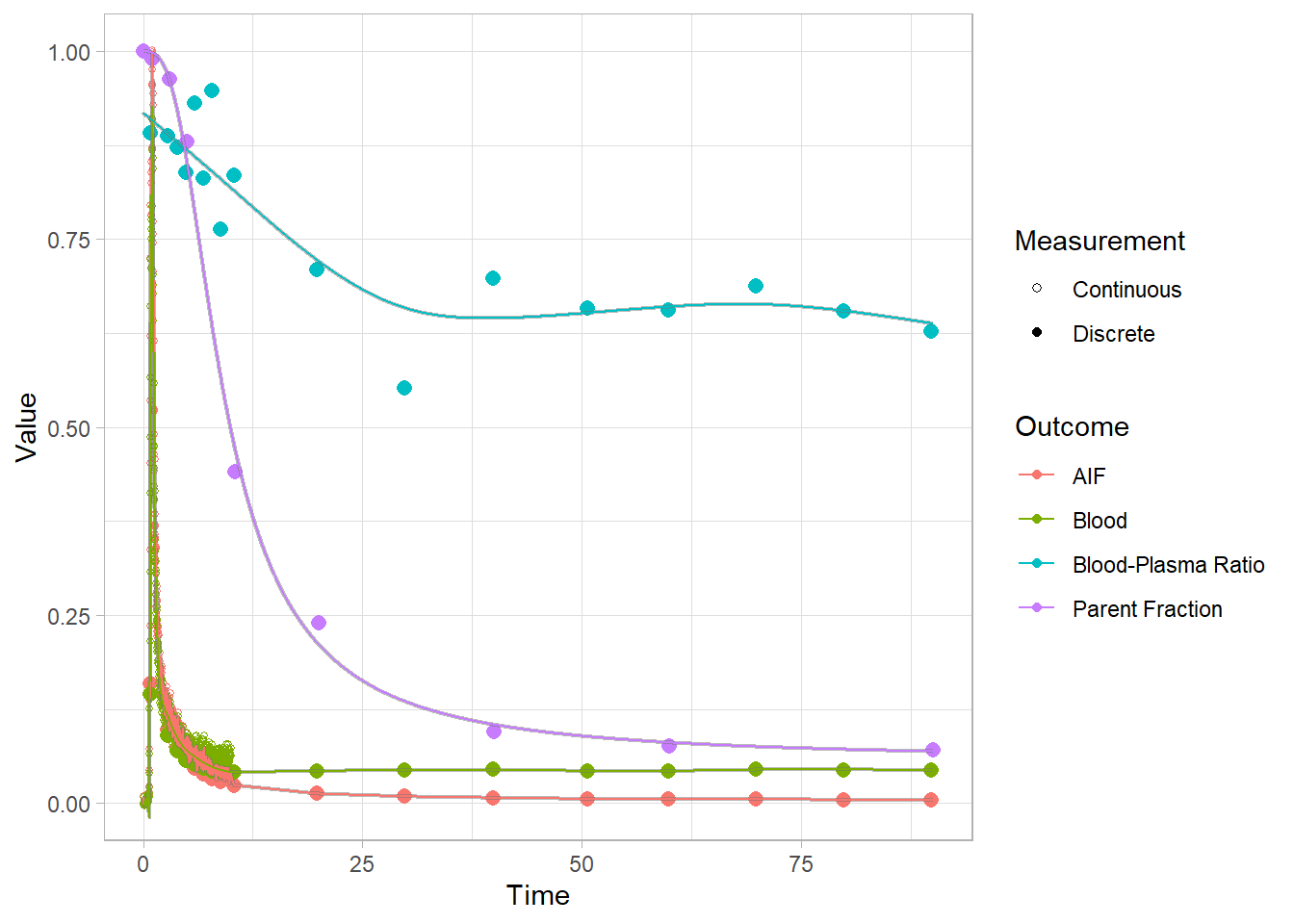
plot(pbr28$blooddata[[2]])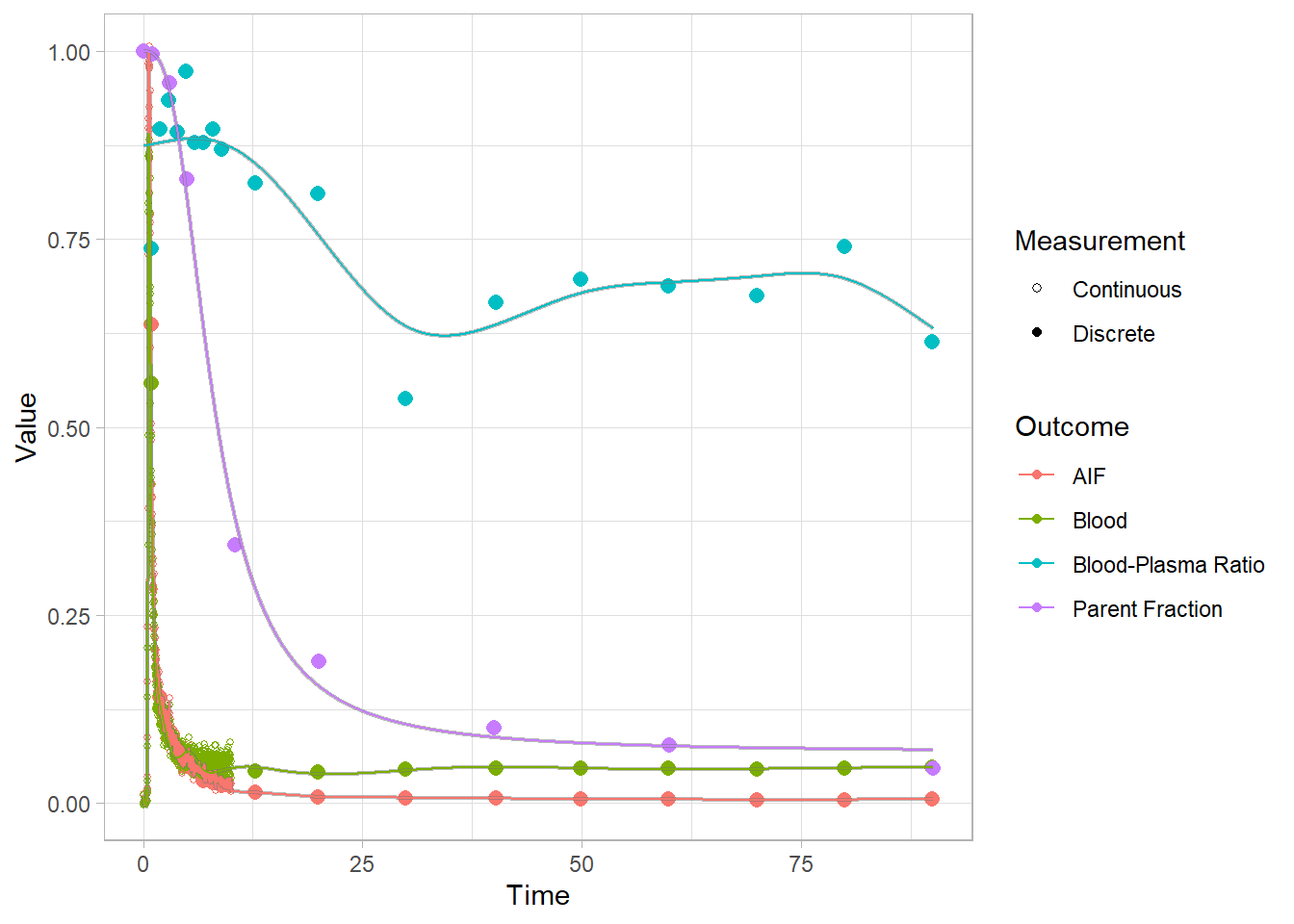
plot(pbr28$blooddata[[3]])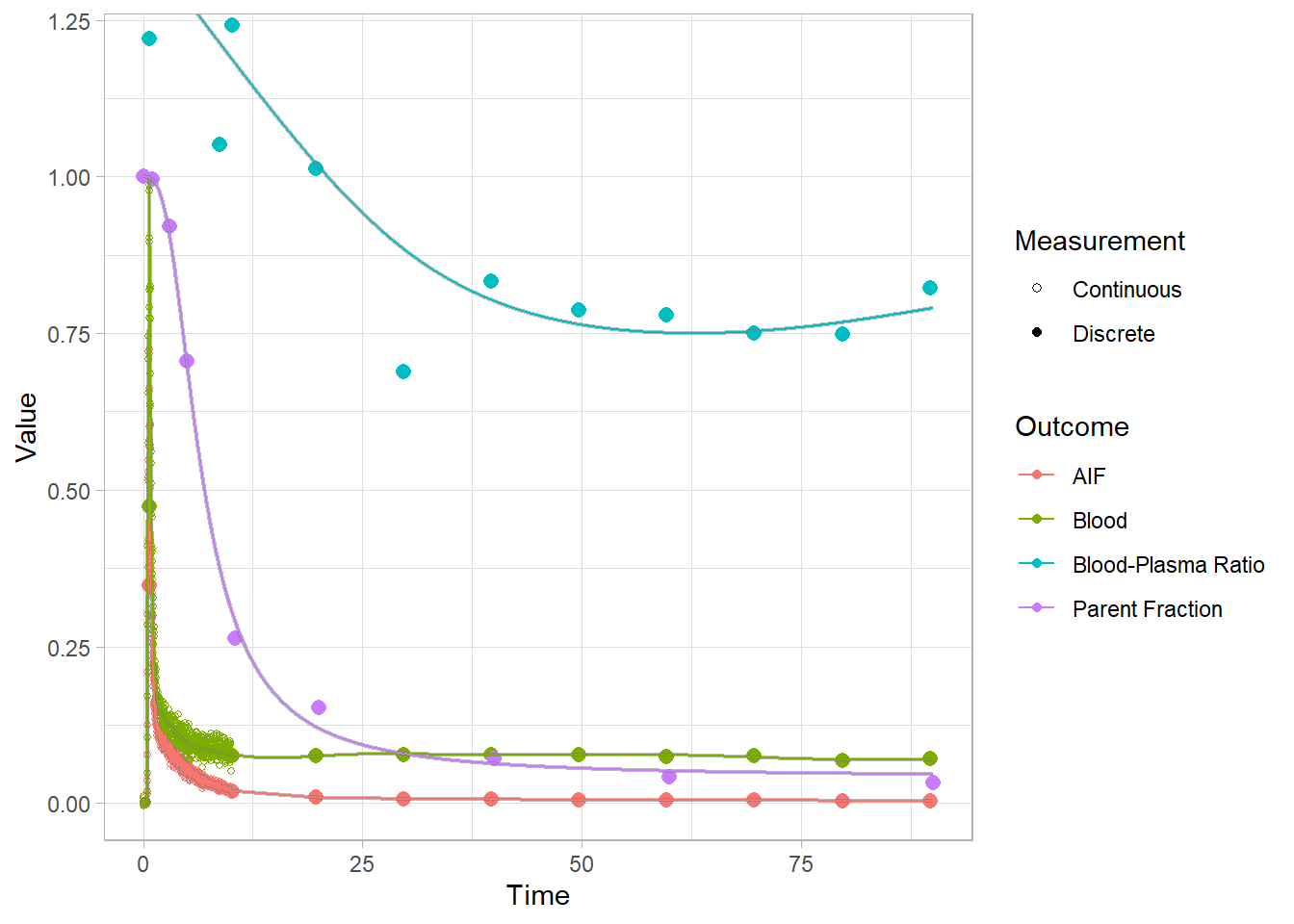
And let’s look at the first 10 minutes to get a better idea of what’s going on.
plot(pbr28$blooddata[[1]]) + xlim(c(0,10))## Warning: Removed 32 rows containing missing values (geom_point).## Warning: Removed 21332 row(s) containing missing values (geom_path).
## Warning: Removed 21332 row(s) containing missing values (geom_path).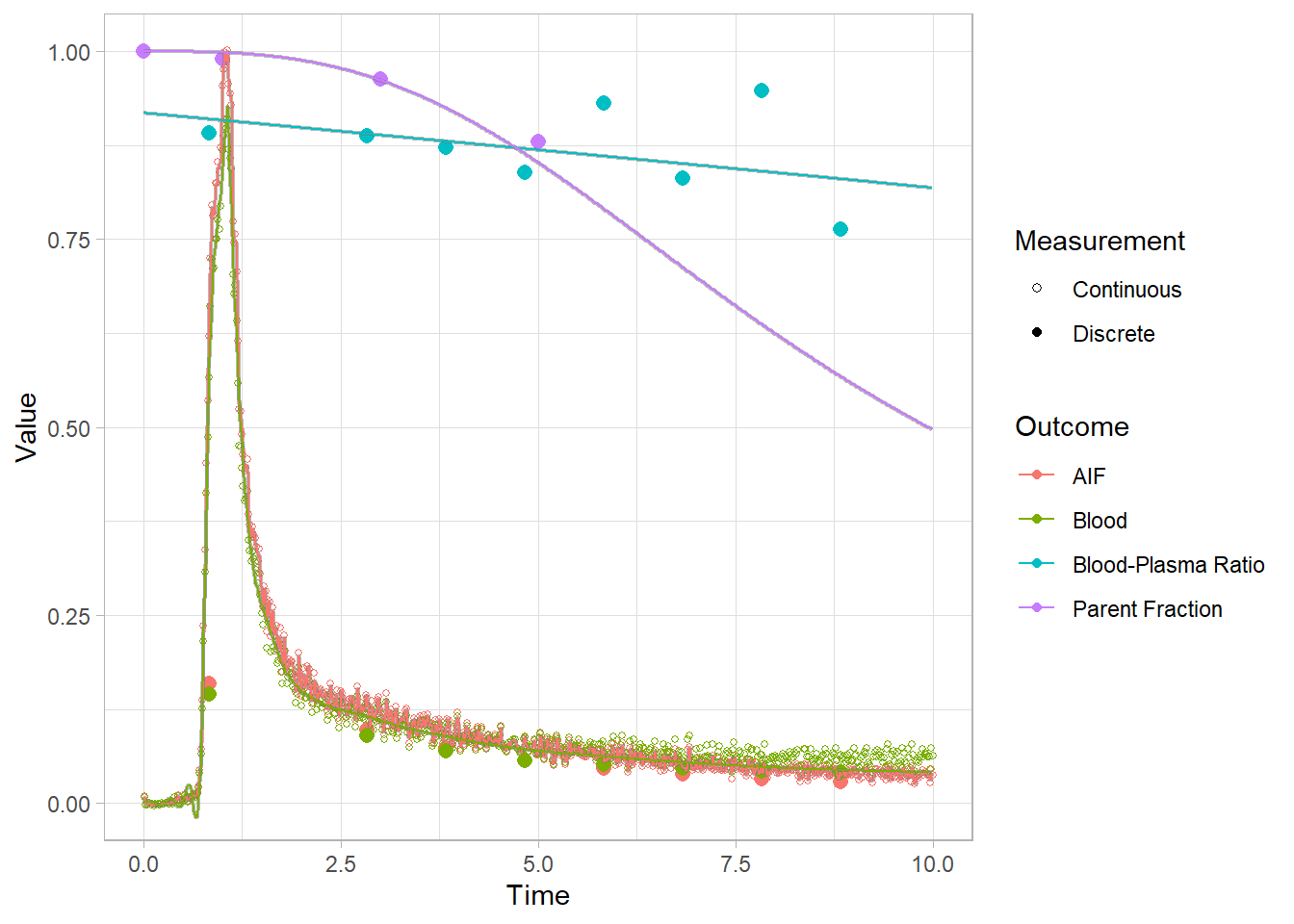
plot(pbr28$blooddata[[2]]) + xlim(c(0,10))## Warning: Removed 32 rows containing missing values (geom_point).
## Warning: Removed 21332 row(s) containing missing values (geom_path).
## Warning: Removed 21332 row(s) containing missing values (geom_path).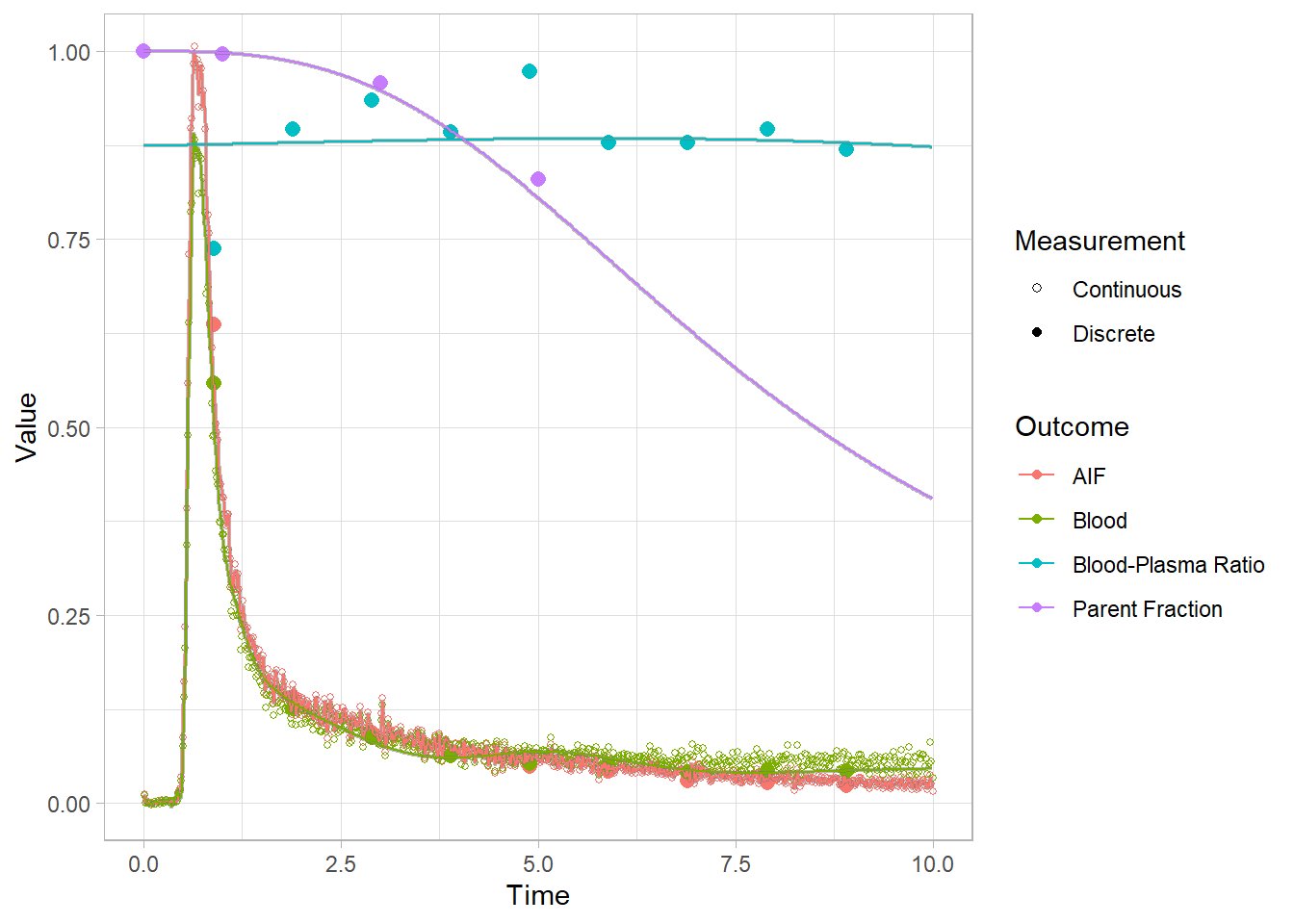
plot(pbr28$blooddata[[3]]) + xlim(c(0,10))## Warning: Removed 32 rows containing missing values (geom_point).
## Warning: Removed 21332 row(s) containing missing values (geom_path).
## Warning: Removed 21332 row(s) containing missing values (geom_path).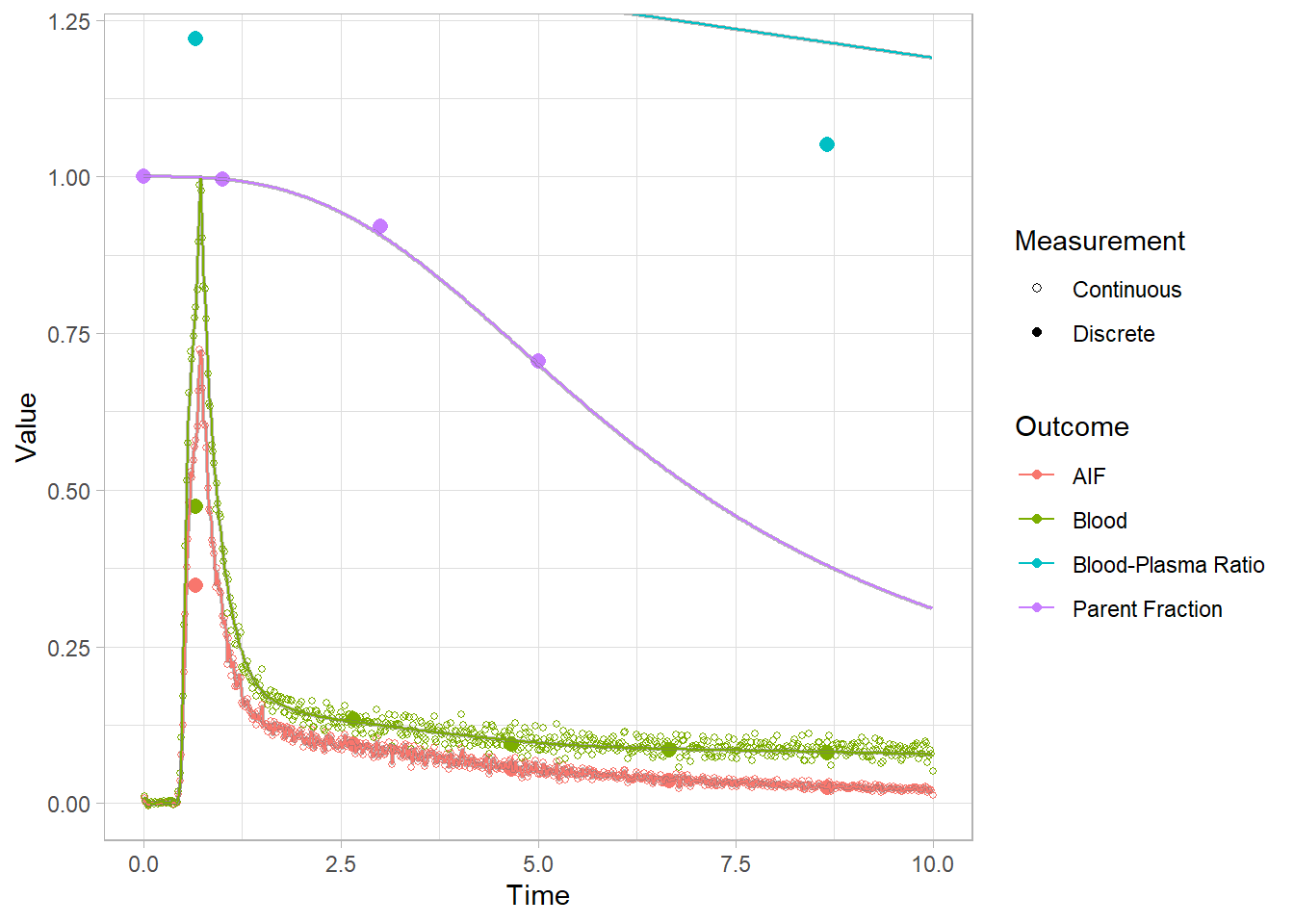
AIF
Now, when it comes to the AIF, it is again important to remember just what our “data” is. We have blood data, which gets multiplied by the blood-to-plasma ratio to get the plasma data. Then we have to multiply this through the parent fraction to get the metabolite-corrected AIF. So, modelling the previous curves has served as preprocessing for getting our AIF data to look as good as we can get it. And now we’re going to model the result of those steps.
Let’s just take a look to see the effect of our processing. We saved a single blooddata object earlier before we did any processing, and never added any fits besides to plot it. So let’s compare the same set before and after modelling. Look especially at the AIF data, and we’ll zoom in on the first 12 minutes to compare.
plot(blooddata) + xlim(c(0,12)) + labs(title="Before Modelling")## Warning: Removed 28 rows containing missing values (geom_point).## Warning: Removed 20801 row(s) containing missing values (geom_path).
## Warning: Removed 20801 row(s) containing missing values (geom_path).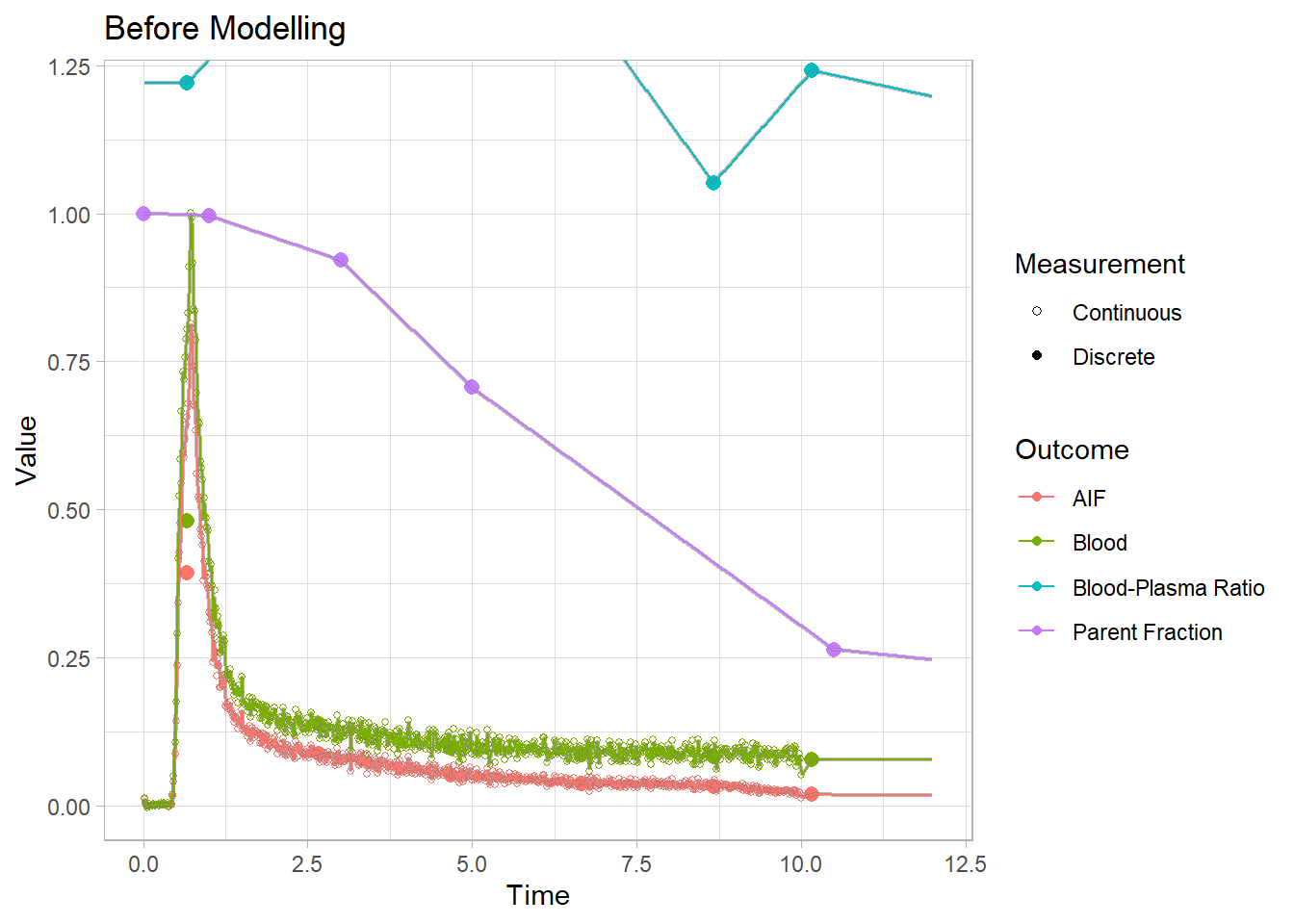
plot(pbr28$blooddata[[1]]) + xlim(c(0,12)) + labs(title="After Modelling")## Warning: Removed 28 rows containing missing values (geom_point).## Warning: Removed 20800 row(s) containing missing values (geom_path).
## Warning: Removed 20800 row(s) containing missing values (geom_path).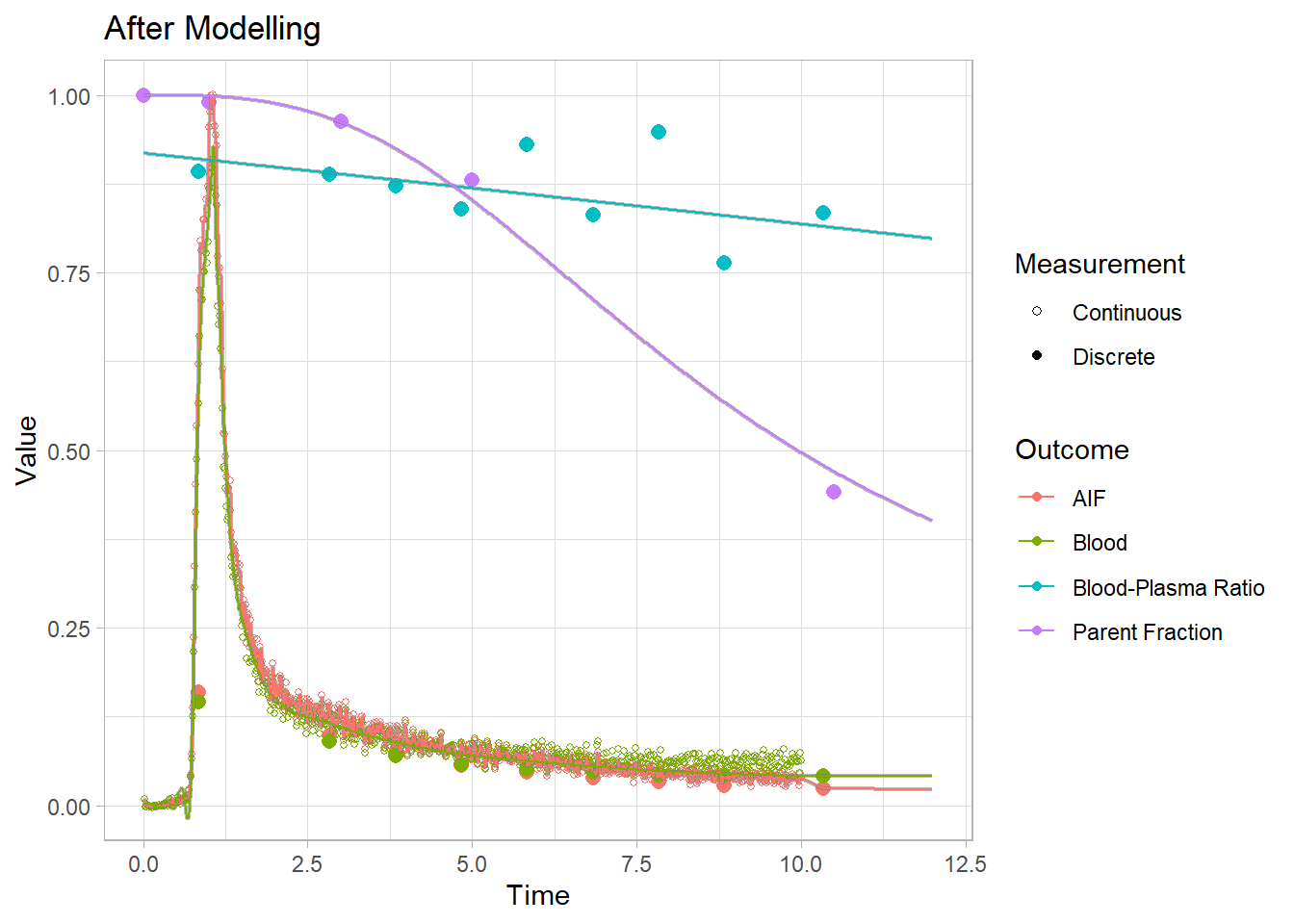
Note: At this point, it’s worth mentioning that I did the blood modelling above mostly to make this point more clear. If you are going to model your AIF, I would advise either not modelling the blood, or else modelling it after the AIF. Because, by doing it here, our AIF is extremely dependent on the blood model. And we would prefer to have modelled a slightly rawer version of that data to get a better fit.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(aifdat = map(blooddata,
~bd_extract(.x, output = "AIF")),
aifmodel = map(aifdat,
~blmod_exp(.x$time, .x$aif, Method = .x$Method, )),
blooddata = map2(blooddata, aifmodel,
~bd_addfit(.x, .y, modeltype = "AIF"))) %>%
select(-aifdat, -aifmodel)I’ve not run this above, because when I do, we hit upon errors because the model fails to converge. This is the result of having modelled the blood first: our model is not managing to fit to the data. Let’s set the blood to simply be interpolated for now, we’ll model the AIF using the raw blood, and then we’ll go back to the smoothed blood later.
First, let’s have a look at what the blooddata object looks like here.
pbr28$blooddata[[1]]$Models$Blood$Method## [1] "fit"Ok, so we’re now using a “fit” instead of an “interp”. Let’s change this to an “interp” again and try to model the data again. It’s worth noting here that each time we extract AIF data, it is regenerated with the current configurations.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(blooddata=map(blooddata, ~modify_in(.x,
c("Models", "Blood", "Method"),
~ "interp"))) %>%
mutate(aifdat = map(blooddata,
~bd_extract(.x, output = "AIF")),
aifmodel = map(aifdat,
~blmod_exp(.x$time, .x$aif, Method = .x$Method, )),
blooddata = map2(blooddata, aifmodel,
~bd_addfit(.x, .y, modeltype = "AIF"))) %>%
select(-aifdat, -aifmodel)And now it works!
It’s also worth noting here, that the bi- or tri-exponential model (you can do either with the blmod_exp() function) for blood curves can be quite tricky to get right. This model is not identifiable, as it is a sum of two or three exponential decays, and one decay can be accommodated by another. So, inside the function, we have a function to select starting parameters, and the starting parameters really are at the core of getting this model to work (trust me! I’ve spent far too long with this model). If your fit does not work, I would advise messing around with the expdecay_props first.
These values defines the fractions of the curve for which the starting parameters are estimated for. So, first, we estimate the third exponential by assuming it applies mostly to the end of the curve, and then we shave this exponential away to estimate the second exponential, and we assume that this exponential primarily applies to a different fraction of the curve. My assumption has been that there is an optimal set of these values for each tracer, but this might not be the case. In any case, if your model is not working, this is the first place to start experimenting. There are also a lot of different configurations of this model. Look into the documentation to understand them better if you are interested, but the default configuration is usually the one to go for in my opinion.
And let’s check how the plots look. Now we’re looking at the red curves.
plot(pbr28$blooddata[[1]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).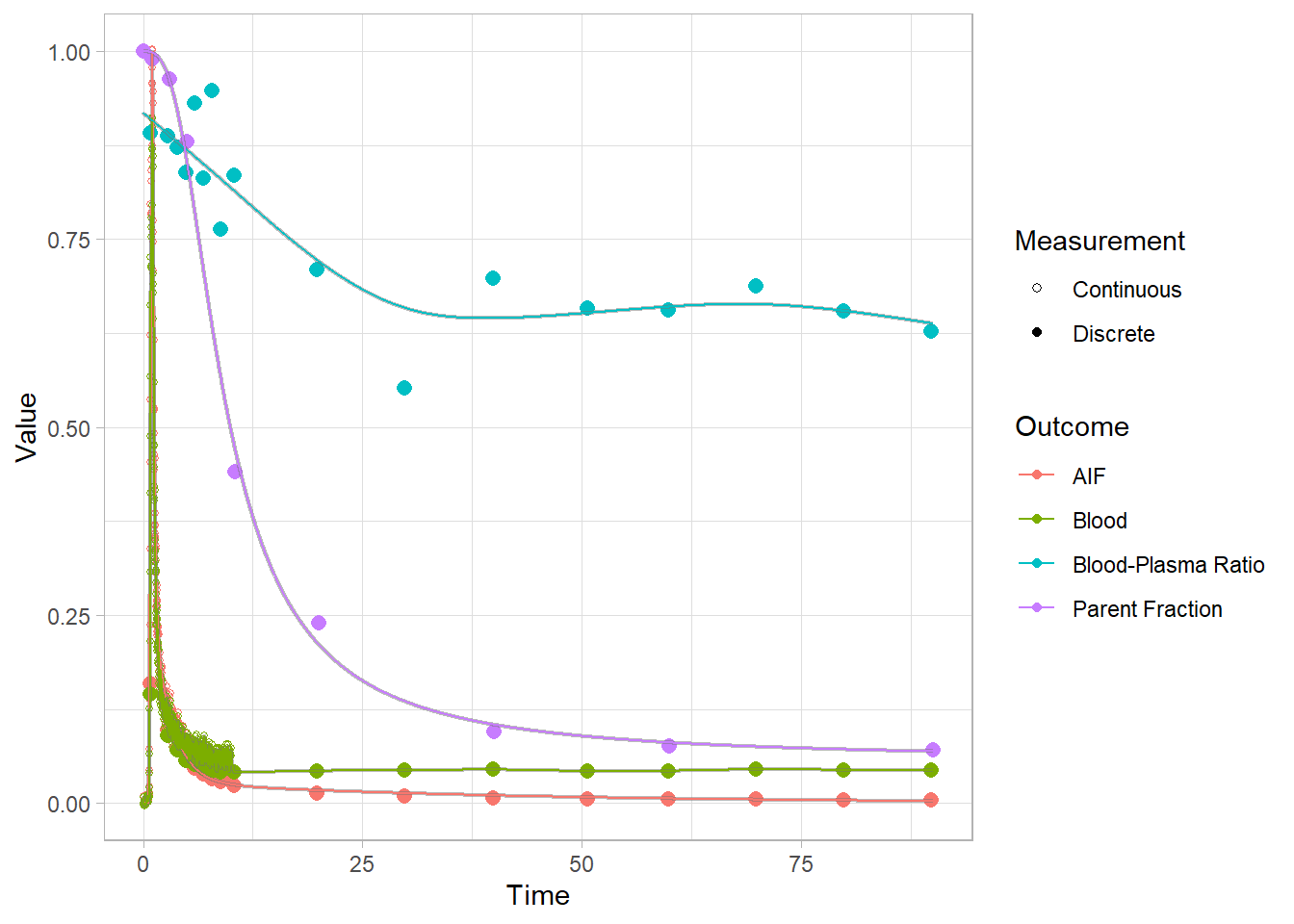
plot(pbr28$blooddata[[2]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).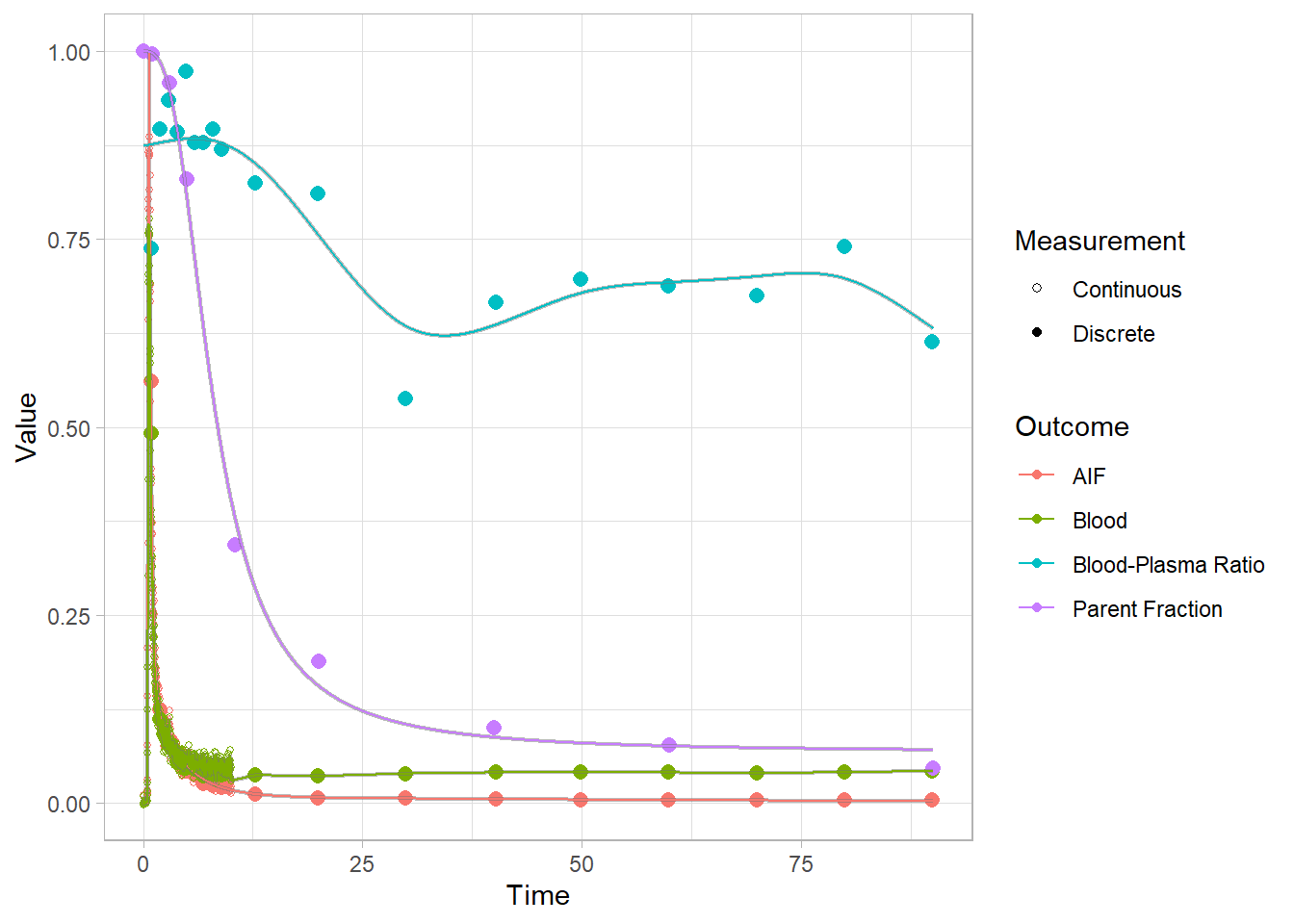
plot(pbr28$blooddata[[3]])## Warning: Removed 1 row(s) containing missing values (geom_path).
## Warning: Removed 1 row(s) containing missing values (geom_path).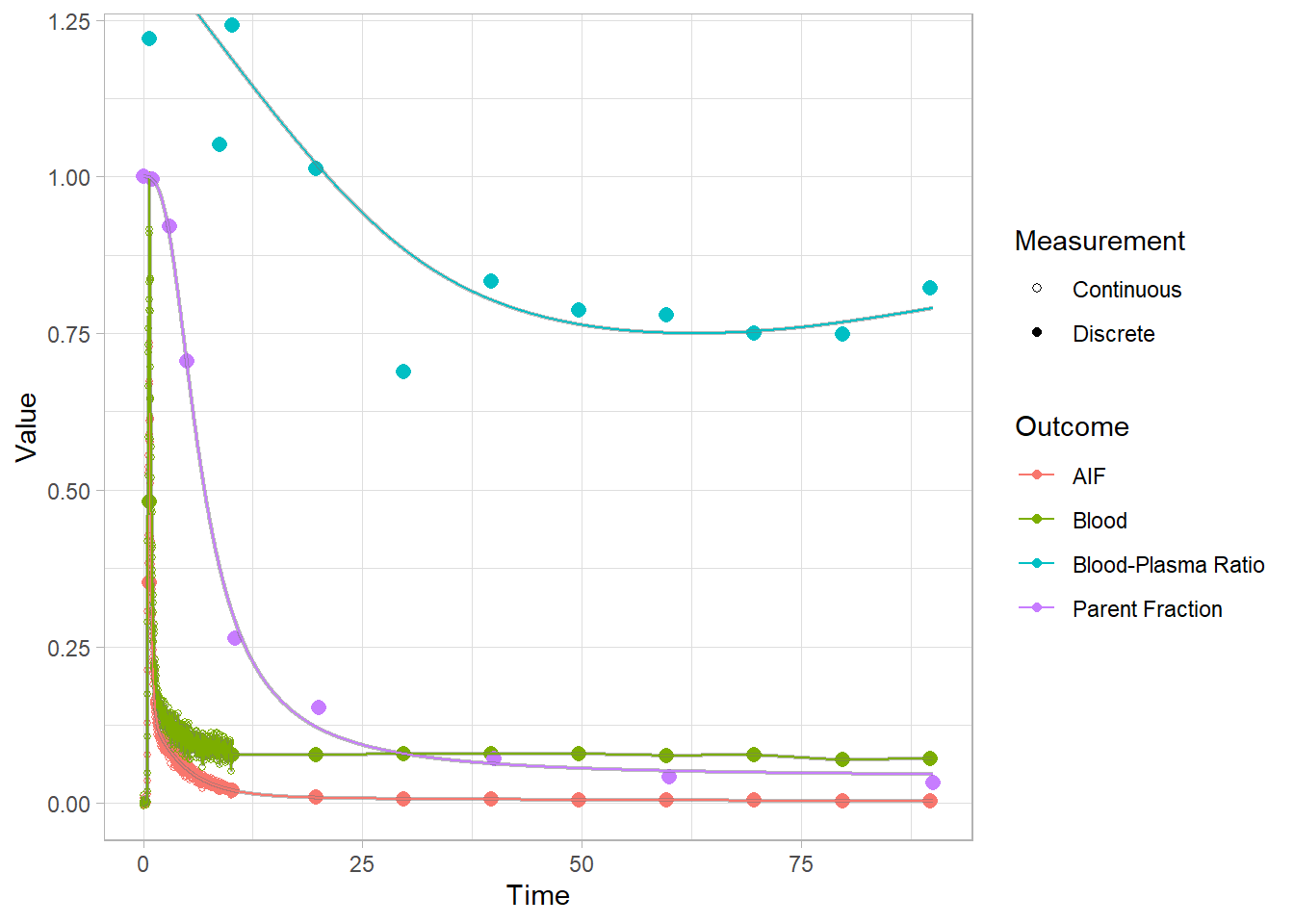
We can’t see that much here: let’s take a look at the start to get a better idea.
plot(pbr28$blooddata[[1]]) + xlim(c(0,5))## Warning: Removed 644 rows containing missing values (geom_point).## Warning: Removed 22665 row(s) containing missing values (geom_path).
## Warning: Removed 22665 row(s) containing missing values (geom_path).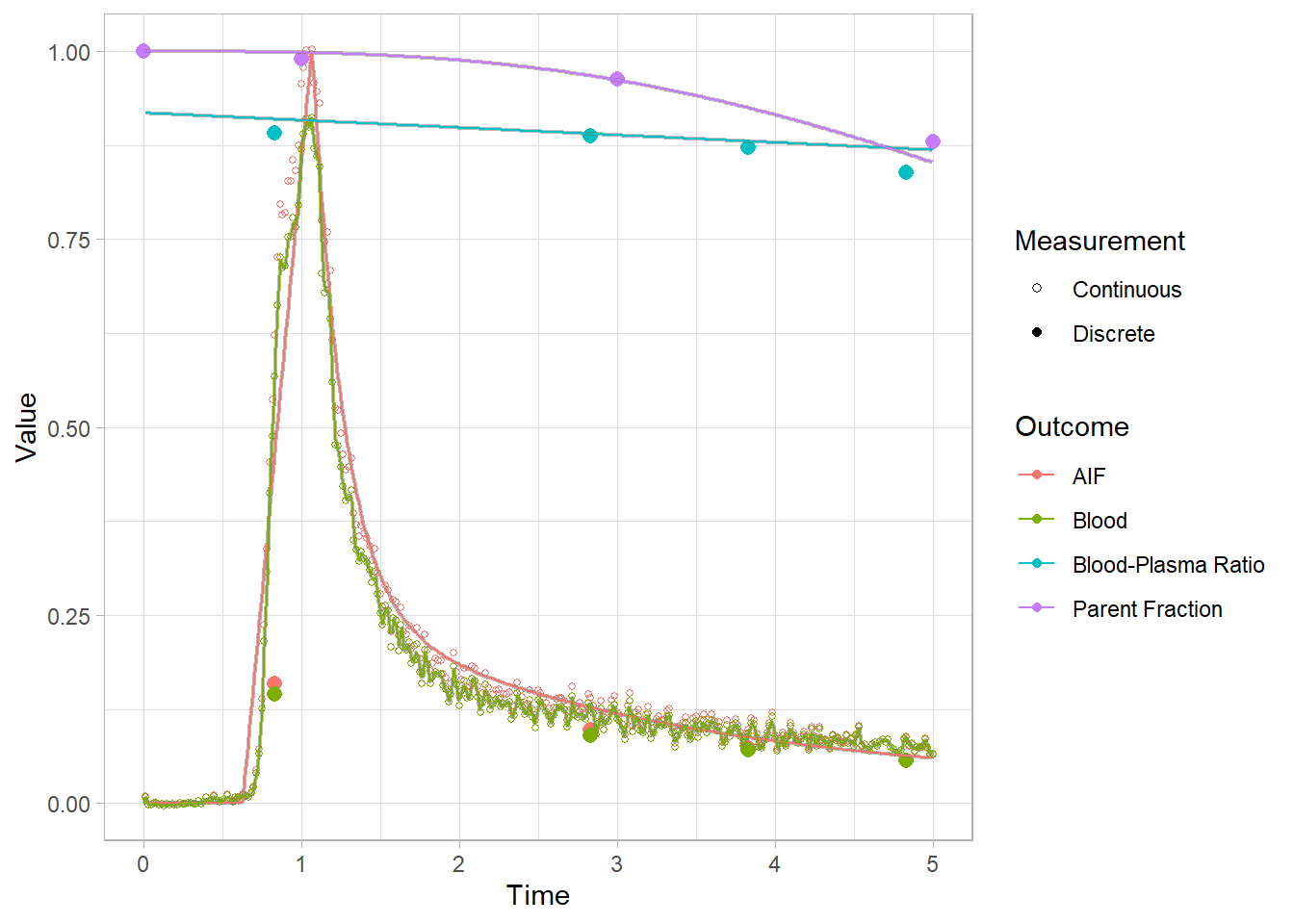
plot(pbr28$blooddata[[2]]) + xlim(c(0,5))## Warning: Removed 644 rows containing missing values (geom_point).
## Warning: Removed 22665 row(s) containing missing values (geom_path).
## Warning: Removed 22665 row(s) containing missing values (geom_path).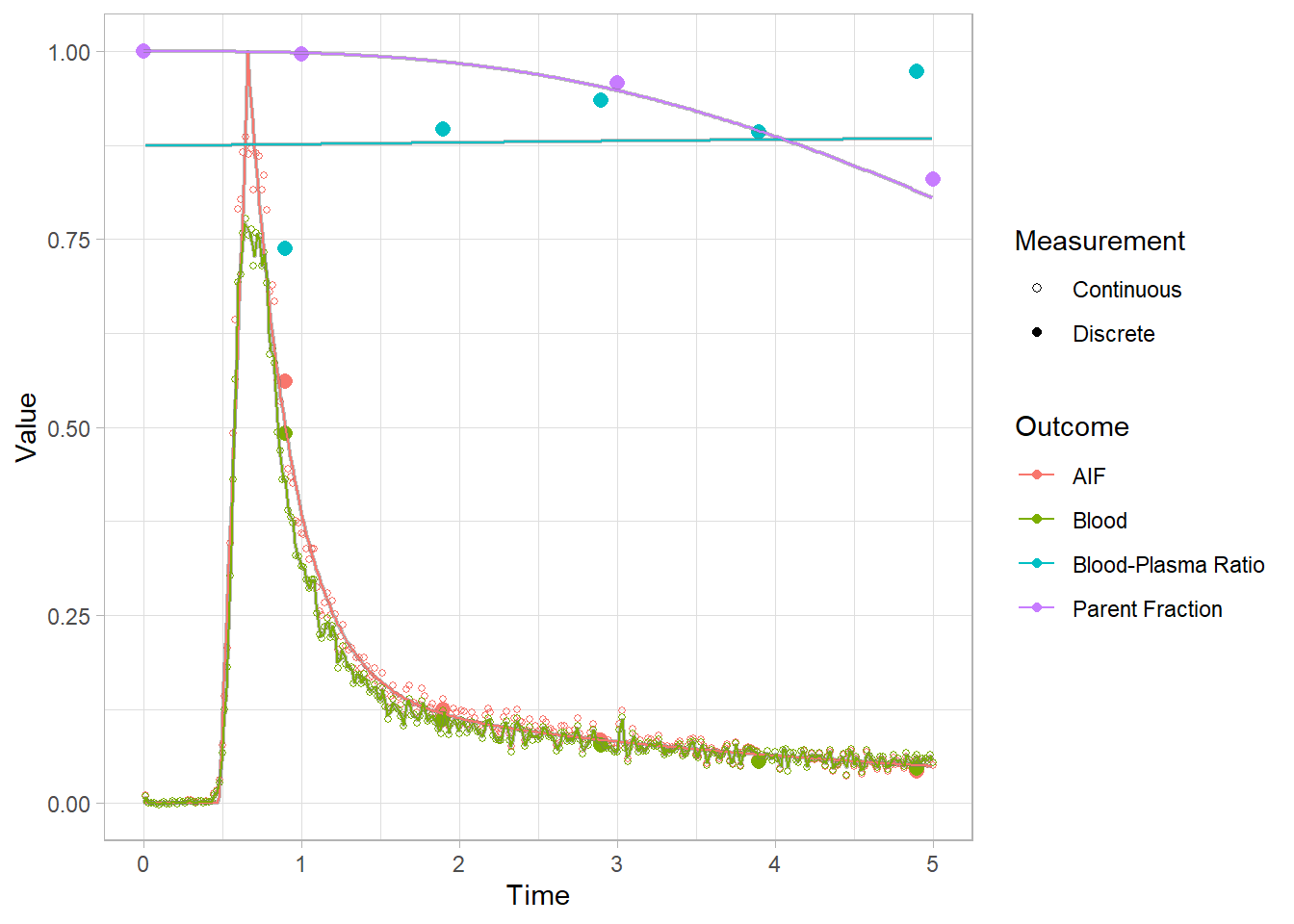
plot(pbr28$blooddata[[3]]) + xlim(c(0,5))## Warning: Removed 638 rows containing missing values (geom_point).
## Warning: Removed 22665 row(s) containing missing values (geom_path).
## Warning: Removed 22665 row(s) containing missing values (geom_path).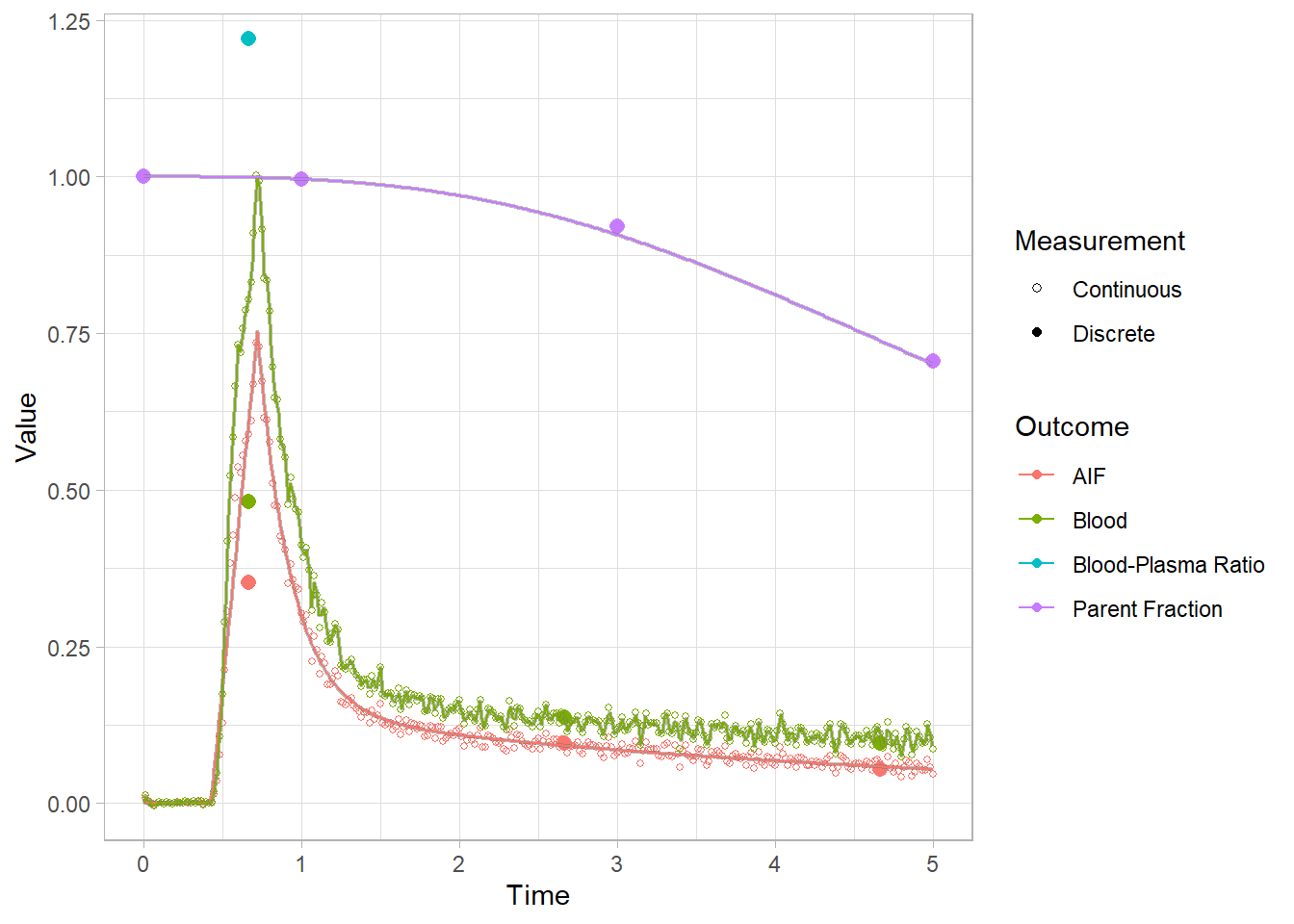
Lookin’ good! Let’s re-smooth our blood curves and move on. Recall that we only changed the method, and didn’t remove the model fit object. So we’ll just change it back to use a fit.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(blooddata=map(blooddata, ~modify_in(.x,
c("Models", "Blood", "Method"),
~ "fit")))And we can take one more peak.
plot(pbr28$blooddata[[1]]) + xlim(c(0,12)) + labs(title="12 minutes")## Warning: Removed 28 rows containing missing values (geom_point).## Warning: Removed 20800 row(s) containing missing values (geom_path).
## Warning: Removed 20800 row(s) containing missing values (geom_path).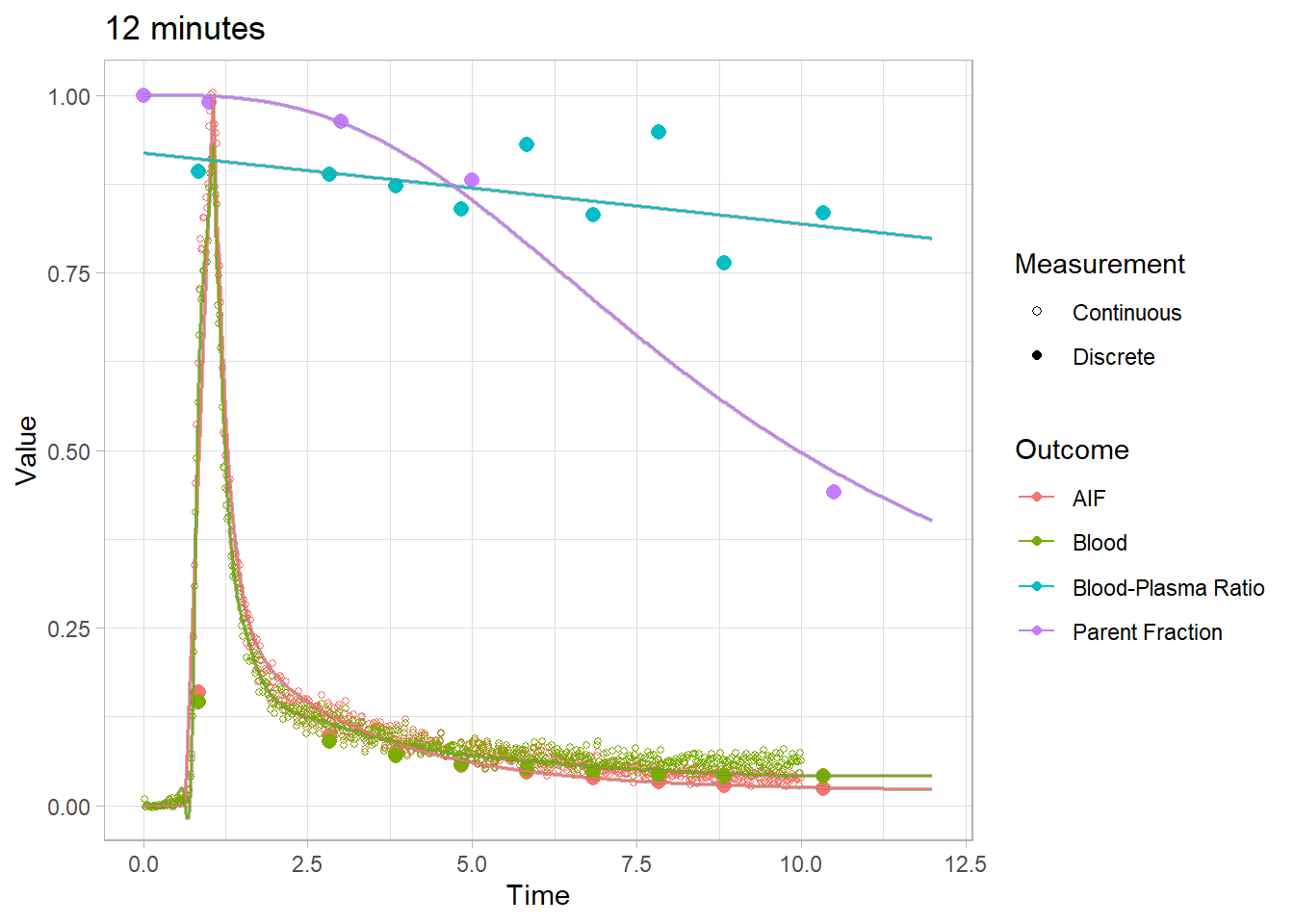
plot(pbr28$blooddata[[1]]) + xlim(c(0,5)) + labs(title="5 minutes")## Warning: Removed 644 rows containing missing values (geom_point).## Warning: Removed 22664 row(s) containing missing values (geom_path).
## Warning: Removed 22664 row(s) containing missing values (geom_path).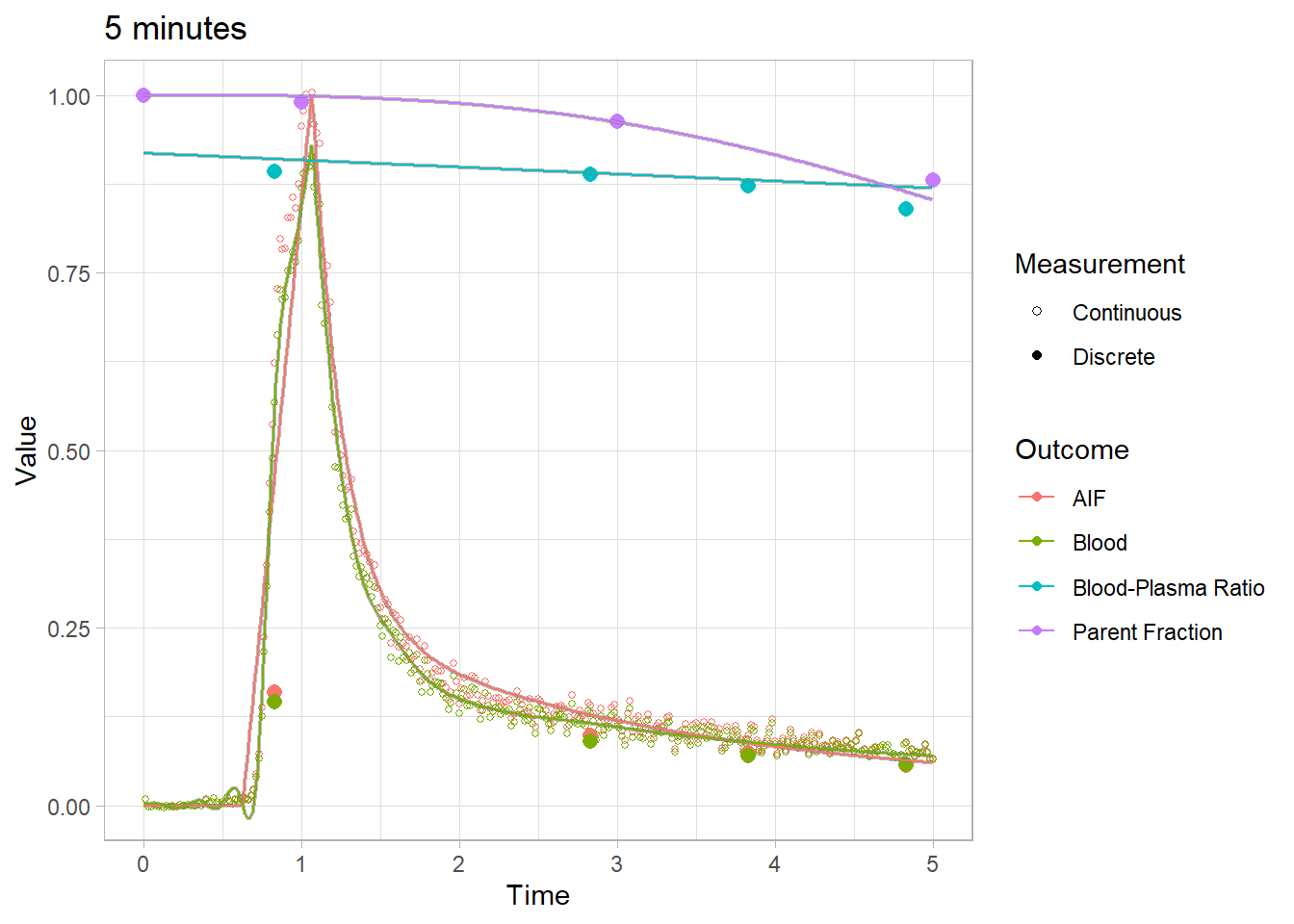
Interpolating the blood data for kinetic modelling
Now, we want to extract the interpolated curves for the purpose of using this in the kinetic models. This means, in kinfitr, extracting an input object from a blooddata object.
pbr28 <- pbr28 %>%
group_by(PET) %>%
mutate(input = map(blooddata, bd_create_input))Done! Now we can get to modelling. See Part 2.
comments powered by Disqus